


Мышечная дистрофия Дюшенна
Прогрессирующие мышечные дистрофии. Клиническая картина.
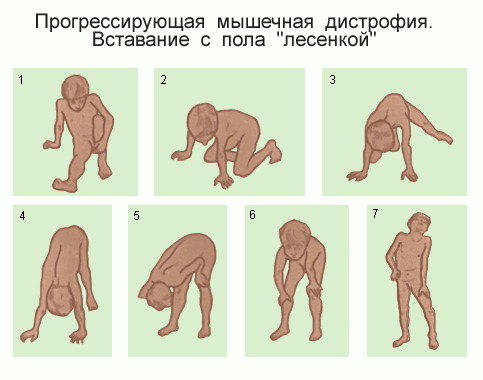 Характерными симптомами ДМП (Дистрофия мышечная прогрессирующая) являются мышечная слабость и атрофия мышц, которые могут проявляться в различные возрастные периоды, но чаще развиваются в детском и юношеском возрасте. Дети поздно начинают ходить, быстро утомляются, неуклюжи в ходьбе, спотыкаются при беге, часто падают, с трудом поднимаются по лестнице. Двигательные нарушения постепенно прогрессируют. Возникает миопатическая утиная походка. В случае поражения мышц тазового пояса и конечностей затруднен переход из горизонтального положения в вертикальное; при поражении дистальных групп мышц ног появляется петушиная походка. Стойкость и нарастание двигательных нарушений позволяют диагностировать миодистрофию уже на ранних стадиях заболеваниях. При обследовании больного обнаруживают генерализованную или локальную атрофию мышц. Локальная атрофия мышц выявляется лишь на ранних стадиях заболевания, по мере прогрессирования патологического процесса атрофия мышц приобретает генерализованный характер вплоть до мышечной кахексии. Атрофированные мышцы истончены, дряблые при пальпации, однако следует отметить, что наряду с атрофией мышц выявляется псевдогипертрофия (замещение атрофированных мышц жировой клетчаткой, соединительной тканью). Миодистрофический процесс сопровождается поражением соединительной ткани, миосклерозом, развитием сухожильно-связочных ретракций, ограничением объема движений в суставах, укорочением пяточного (ахиллова) сухожилия, контрактурами. Одновременно с развитием мышечных атрофий снижаются сухожильные рефлексы, в первую очередь коленные.
Характерными симптомами ДМП (Дистрофия мышечная прогрессирующая) являются мышечная слабость и атрофия мышц, которые могут проявляться в различные возрастные периоды, но чаще развиваются в детском и юношеском возрасте. Дети поздно начинают ходить, быстро утомляются, неуклюжи в ходьбе, спотыкаются при беге, часто падают, с трудом поднимаются по лестнице. Двигательные нарушения постепенно прогрессируют. Возникает миопатическая утиная походка. В случае поражения мышц тазового пояса и конечностей затруднен переход из горизонтального положения в вертикальное; при поражении дистальных групп мышц ног появляется петушиная походка. Стойкость и нарастание двигательных нарушений позволяют диагностировать миодистрофию уже на ранних стадиях заболеваниях. При обследовании больного обнаруживают генерализованную или локальную атрофию мышц. Локальная атрофия мышц выявляется лишь на ранних стадиях заболевания, по мере прогрессирования патологического процесса атрофия мышц приобретает генерализованный характер вплоть до мышечной кахексии. Атрофированные мышцы истончены, дряблые при пальпации, однако следует отметить, что наряду с атрофией мышц выявляется псевдогипертрофия (замещение атрофированных мышц жировой клетчаткой, соединительной тканью). Миодистрофический процесс сопровождается поражением соединительной ткани, миосклерозом, развитием сухожильно-связочных ретракций, ограничением объема движений в суставах, укорочением пяточного (ахиллова) сухожилия, контрактурами. Одновременно с развитием мышечных атрофий снижаются сухожильные рефлексы, в первую очередь коленные.
- Псевдогипертрофическая злокачественная миодистрофия Дюшенна проявляется в возрасте 2—5 лет. Обездвиженность больных, как правило, наступает в возрасте 14—15 лет, смерть наступает в возрасте 15—18 лет. Характерны поясно-конечностная атрофия мышц, преимущественно мышц тазового пояса и бедер, истинная гипертрофия или псевдогипертрофия икроножных мышц, ранние сухожильно-связочные ретракции (укорочение сухожилий и связок), контрактуры крупных суставов. Коленные рефлексы рано исчезают, ахилловы рефлексы сохраняются. Особенностями этой формы Д. м. п. являются сопутствующая поражению мышц умственная отсталость, снижение интеллекта, адипозогенитальный синдром, остеопороз и истончение кортикального вещества костей, кардиомиопатия, легочно-сердечная недостаточность. Уже в ранних стадиях заболевания обнаруживают креатинурию, гипераминоацидурию, повышение альдолаз, трансаминаз (особенно аланиновой) и специфического фермента мышечной ткани — креатинфосфокиназы. Нарушения всех видов обмена веществ (углеводного, жирового, белкового), гипераминоацидурия, гиперферментурия, пентозурия, креатинурия могут наблюдаться и при других формах нервно-мышечных заболеваний. Однако при миодистрофии Дюшенна биохимические изменения выражены в большей степени, что является дополнительным критерием при оценке тяжести заболевания. Эти изменения служат биохимическими маркерами при выявлении гетерозиготных носителей. Миодистрофия Дюшенна передается по рецессивному типу, сцепленному с полом. У матери — носительницы мутантного гена обнаруживают малые признаки болезни: уплотнение икроножных мышц, мышечную слабость при физической нагрузке, миодистрофические изменения при электромиографии, гиперферментемию. Наряду с классическим рецессивным типом наследования встречаются отклонения в типе наследования, например существуют семьи, где все мальчики страдают этим заболеванием, а также семьи, где заболевание встречается у девочек.
- Псевдогипертрофическая доброкачественная миодистрофия Беккера — Кинера начинается в возрасте 10—15 лет. Поражаются мышцы проксимальных отделов конечностей, тазового и плечевого пояса, Электромиографические, биохимические и патоморфологические изменения умеренно выражены, отмечается изменение цветового зрения. Длительное время у больных сохраняются трудоспособность, возможность самостоятельного передвижения, интеллект; кардиомиопатия выражена умеренно.
- Миодистрофия Дрейфуса — Хогана относится к редким Х-хромосомным формам Д. м. п. Заболевание начинается в возрасте 4—5 лет. Характеризуется проксимальным тетрапарезом, ранним развитием ретракции и контрактур, медленным прогрессированием. Причиной смерти, наступающей в возрасте 30—40 лет, обычно является сердечная недостаточность.
- Юношеская псевдогипертрофия Мэбри начинается в возрасте 11—13 лет. Характерные ее признаки: выраженные псевдогипертрофии, умеренный проксимальный тетрапарез, сохранность интеллекта, отсутствие ретракции и контрактур.
- Миодистрофия Роттауфа — Мортье начинается в возрасте 8—9 лет, отличается выраженным миосклерозом, развитием сухожильных ретракции и контрактур в локтевых, голеностопных суставах, ригидности позвоночника. Мышечные атрофии преобладают в лопаточно-плечевой области и в дистальных отделах конечностей. Псевдогипертрофии отсутствуют, интеллект сохранен. Течение заболевания медленное, больные длительно сохраняют подвижность и обслуживают себя.
- Поясно-конечностная юношеская миодистрофия Эрба — Рота. Тип наследования аутосомно-рецессивный, имеются спорадические случаи. Заболевание проявляется в возрасте 13—16 лет, однако первые его признаки могут наблюдаться в раннем детском возрасте (дистрофия Лейдена). Характерными симптомами являются слабость и атрофия мышц тазового пояса и бедер, мышц живота и туловища, что проявляется гиперлордозом позвоночника, выпячиванием живота, утиной походкой, затруднением при переходе из горизонтального положения в вертикальное. Генерализация атрофий происходит по восходящему типу. Псевдогипертрофии икроножных мышц, ретракции и контрактуры выражены умеренно, интеллект сохранен. Кардиомиопатия проявляется в поздних стадиях заболевания. Атрофия дыхательных мышц, деформация грудной клетки и позвоночника приводят к нарушению функции внешнего дыхания, легочно-сердечной недостаточности. Смерть больных обычно наступает от бронхопневмонии. С помощью электромиографии выявляют снижение амплитуды, полифазность биопотенциалов. При биохимическом исследовании обнаруживают гиперферментурию, гипер-миноацидурию, креатинурию, пентодурию и др., но выраженные в меньшей степени, чем у больных миодистрофией Дюшенна. Клиническое обследование членов семьи больного, а также электрофизиологическое и биохимическое исследования позволяют выявить малые признаки болезни.
- Плече-лопаточно-лицевая миодистрофия Ландузи — Дежерина. Тип наследования аутосомно-доминантный, имеются спорадические случаи. Заболевание начинается в возрасте 12—20 лет. Характерны слабость и атрофия мышц лица, плечевого пояса, проксимальных отделов рук, следствием чего являются амимия, отставание лопаток (крыловидные лопатки) при попытке поднять руки вверх, деформация грудной клетки и позвоночника. Генерализация патологического процесса продолжается 10—15 лет, он постепенно распространяется на мышцы тазового пояса, проксимальных и дистальных отделов ног. В некоторых случаях развиваются также атрофии мышц бедер и голеней. Псевдогипертрофия икроножных, дельтовидных, лицевых мышц выражена умеренно. Рефлексы могут быть долгое время сохранены. Течение медленно прогрессирующее, больные длительное время сохраняют трудоспособность. Интенсивные физические нагрузки ведут к быстрому прогрессированию заболевания.
- Дистальная миодистрофия Говерса относится к редким заболеваниям. Тип наследования аутосомно-доминантный, имеются спорадические случаи. Проявляется в возрасте 30—60 лет. Характерными симптомами являются слабость и атрофии мышц голени и стоп, снижение ахилловых и коленных рефлексов. Генерализация процесса с развитием атрофий кистей и проксимальных отделов конечностей происходит в течение 5—10 лет. У больных, как правило, выражена кардиомиопатия, приводящая к летальному исходу.
- Офтальмоплегическая и окулофарингеальная (глазо-глоточная) формы. Тип наследования аутосомно-доминантный, имеются спорадические случаи. Возраст начала заболевания может быть разным. Характерны медленно нарастающая слабость и атрофия глазодвигательных мышц, ограничение движений глазных яблок, опущение верхнего века. Поражение глазных мышц может быть изолированным или сочетаться с атрофией мышц лица, глотки (что приводит к затруднению глотания), поражением мышц конечностей. Течение медленно прогрессирующее.
Диагноз. Д. м. п. может быть поставлен в амбулаторных условиях. Обследование в стационаре необходимо в случаях редких форм или для проведения дифференциального диагноза. Во всех случаях обнаружения у больного мышечной слабости и атрофии, двигательных нарушений прогрессирующего характера необходимо изучить семейный анамнез, обследовать родственников (гетерозиготных носителей) для выявления у них малых признаков заболевания. Дифференциальный диагноз следует проводить с врожденными миопатиями, характерными признаками которых являются диффузная мышечная гипотония, гипотрофия. Д. м. п. следует также дифференцировать с вторичными миопатическими синдромами, которые возникают на фоне воспалительных, сосудистых, токсических, метаболических процессов, или обусловлены воздействием физических факторов.
информация с портала miopatia.ru
Что такое мышечная дистрофия Дюшенна?
Есть много типов мышечной дистрофии, все они вызваны нарушением генов (единиц наследственности, передаваемых от родителей к детям). При мышечной дистрофии Дюшенна (МДД) недостаток белка дистрофина вызывает ухудшение и разрушение мышц, ведущее к прогрессирующему затруднению ходьбы и общей подвижности. МДД является наиболее часто случающейся и одной из наиболее быстро прогрессирующих детских нейромышечных болезней. Этой болезнью болеет приблизительно каждый 3000-ый новорожденный мальчик в мире. МДД болеют только мальчики (за очень редким исключением)…
Как мышечная дистрофия Дюшенна наследуется?
При мышечной дистрофии Дюшенна дефектный ген является Х-сцепленным. Это означает, что этот ген расположен на Х-хромосоме. У женщин две Х-хромосомы, а у мужчин одна Х-хромосома, которую они наследуют от своей матери, и одна Y-хромосома, которую они наследуют от своего отца. Приблизительно в двух третьих случаев дефектный ген передается сыну посредством дефектной Х-хромосомы матери. В этих случаях мать является «носителем», у которого, в большинстве случаев, не проявляются никакие симптомы болезни. Это потому, что этот ген является «рецессивным», а что означает, что ее нормальная Х-хромосома будет доминантной и будет нормально производить дистрофин. Только у очень малого количества носителей наблюдается умеренная степень мышечной слабости, которая обычно ограничивается плечами и бедрами, и такие женщины называются «проявляющимися носителями». Генетическое нарушение могло возникнуть в предыдущем поколении, в котором наблюдалась семейная предрасположенность к этому заболеванию. Однако, приблизительно в одной трети случаев МДД генетическое нарушение возникает в самом мальчике, и тогда оно называется «спонтанной мутацией».
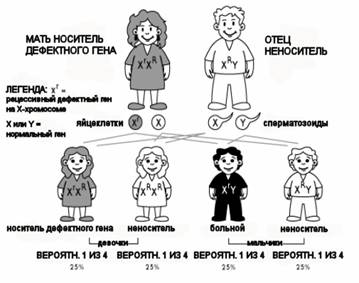
Почему так важно консультирование по вопросам наследственности?
Каждый сын женщины-носителя имеет 50% вероятность унаследовать MДД от дефектной Х-хромосомы его матери, и каждая дочь имеет 50% вероятность стать носителем этой болезни таким же способом. Сразу после диагноза МДД необходимо получить консультацию по вопросам наследственности, а также пройти надлежащее тестирование членам семьи, которые возможно являются носителями. Во время консультации вы получите информацию о последовательности наследственности и об опасности для других членов семьи, а также «прогноз» (возможные последствия болезни). Во время этой консультации также предоставляется информация о диагностическом тестировании, включая предродовое тестирование и тестирование носителя.
Как диагностируется МДД?
Симптомы
Болезнь МДД часто трудно диагностировать, так как симптомы могут быть разными, и если в семье не было этой болезни, наличие МДД может не подозреваться. Довольно обычным считается запаздывание начала ходьбы, когда ребенок делает первые шаги приблизительно в восемнадцать месяцев. При ходьбе больной МДД мальчик может часто падать. Ему часто трудно взбираться по ступенькам, трудно бегать и прыгать, и у него может развиться «утиная» походка. Классическими симптомами является увеличение (гипертрофия) икроножных мышц, которое случается приблизительно в 90% случаев. У него может развиться тенденция ходить на пальцах, что часто сопровождается выпиранием живота и раздвинутыми в коленных суставах ногами, и называется «лордозом». Ему может быть трудно вставать с пола без помощи. Чтобы помочь себе, он может взбираться руками по ногам – это называется «признаком Говерза». Эти симптомы обычно начинают развиваться в возрасте от одного до трех лет и продолжают прогрессировать до тех пор, пока ему не понадобиться кресло-коляска, чаще всего в возрасте от восьми до двенадцати.
Анализ креатинфосфокиназы
Лабораторное тестирование МДД начинается с анализа мышечного энзима, называемого креатинфосфокиназой. Из-за недостатка дистрофина в волокнах мышцы креатинфосфокиназа просачивается из поврежденной мышцы и появляется в крови в больших количествах. Анализ крови может показать уровень креатинфосфокиназы, превышающий нормальный уровень в 50 – 100 раз. Хотя содержание этого энзима часто слегка повышенное при дистрофии других типов (включая ассоциативную мышечную дистрофию Беккера), при МДД его содержание намного выше. Приблизительно у 70% носителей МДД уровень креатинфосфокиназы также будет слегка повышенный. Поэтому высокий уровень креатинфосфокиназы свидетельствует о том, что сами мышцы являются вероятной причиной слабости, но не говорит нам со 100% гарантией, какая мышечная болезнь это может быть.
Изучение ДНК
В настоящее время, для того чтобы установить точный диагноз, изучение ДНК ведется с применением новых технологий. Гены состоят из сегментов ДНК (дезоксирибонуклеиновой кислоты), и соответствующие части этого генетического материала могут быть изучены при помощи микроскопа. Аномалии, вызывающие МДД, могут быть трех типов: удаление (отсутствующие части), дублирование (дополнительные части) или точечная мутация (измененные части). Изучение ДНК часто занимает много времени и трудно с технической стороны и, в зависимости от генетического дефекта, могут давать неопределенные результаты. В некоторых случаях эти изучения могут дать точную информацию о генетической аномалии, вызывающей МДД, а в других случаях аномалию невозможно точно определить. Это также относится и к диагностике женщин-носителей. Изучение ДНК может также проводиться до родов в неродившемся ребенке, если в семье было это заболевание.
Биопсия мышц
Если изучение ДНК не дает ясной картины, может потребоваться биопсия мышц. Маленькая частица мышечной ткани, обычно из бедра, извлекается при помощи иглы. Используя специальный метод окрашивания в лаборатории, мышечная ткань изучается при помощи микроскопа на наличие дистрофина. При МДД анализ показывает отсутствие дистрофина, в то время как при родственной болезни мышечной дистрофии Беккера небольшое количество дистрофина присутствует. Поэтому анализ биопсии мышц необходим для установления точного анализа в тех случаях, когда неизвестно, болел ли кто в семье этой болезнью, или когда анализ ДНК не дал определенных результатов.
Только две болезни могут вызвать трудности при диагностировании МДД: мышечная дистрофия Беккера и конечностно-поясная мышечная дистрофия. Вышеупомянутые анализы, в особенности биопсия мышц, могут различать эти болезни.
Вылечивается ли МДД?
В настоящее время МДД не вылечивается, но по всему миру продолжаются широкомасштабные исследования в этой области. Исследователи значительно продвинулись в понимании МДД и продолжают поиски лечения. Некоторыми областями, на которых исследования сконцентрированы в настоящий момент, являются:
Заместительная генная терапия, целью которой является производство синтетического гена, который может быть введен в тело, заменяя процесс производства недостающего белка дистрофина. В настоящее время необходимо преодолеть несколько трудностей, прежде чем эта терапия может быть использована для лечения МДД.
Трансплантация миобласта – при этой процедуре клетки донора вводятся в поврежденную мышцу в надежде на то, что они сольются с больной мышцей и создадут несколько нормальных волокон мышцы, которые затем будут производить ген дистрофина. Это лечение пока не доказало свою эффективность.
Появляются ли другие нарушения при МДД?
Нарушение сердечной деятельности
Во многих случаях мышечной дистрофии сердечная ткань может быть повреждена одним из двух способов.
Во-первых, может быть повреждена проводящая ткань, что приводит к ненормальному сердечному ритму (сердце бьется слишком быстро или слишком медленно). Это может привести к появлению таких симптомов, как приступы головокружения, учащенное сердцебиение или временная потеря сознания. Учащенный ритм можно вылечить и предупредить при помощи медикаментов. Замедленный ритм обычно вызывается блокадой сердца и для его лечения может быть необходимо введение кардиостимулятора (делается небольшая операция под местной анестезией).
Во-вторых, может возникнуть нарушение перекачивающей функции сердца, называемое кардиомиопатией, и могут появиться такие симптомы, как одышка. Появление кардиомиопатии более вероятно у людей с МД Дюшенна или Беккера. Диагностирование выполняется при помощи простой проверки сердца ультразвуком, называемой эхокардиограммой (ЭХО), которая может делаться несколько раз с целью наблюдения за действием лечения и прогрессированием болезни. Для улучшения перекачивающей функции сердца используются медикаменты.
Несмотря на то, что серьезные осложнения случаются у немногих, всем больным МДД необходимо регулярно проверять работу сердца при помощи электрокардиограммы (ЭКГ) и проходить медицинские проверки. У женщин-носителей, не показывающих признаков (или показывающих очень слабые признаки) мышечной слабости, может быть небольшая степень кардиомиопатии, и они также должны проходить регулярные проверки.
Нарушение познавательных способности
У большинства (около 65%) больных МДД нормальные интеллектуальные способности и они демонстрируют средний интеллект или выше среднего. Однако, приблизительно у 35% мальчиков, страдающих МДД, наблюдаются нарушения интеллектуальных способностей или трудности с обучением. В этих случаях считается, что у таких больных затруднено получение информации, хранение этой информации и ее последующее отыскивание. Когда такие трудности возникают, считается, что это связано с изменением распределения дистрофина в мозгу. Трудности с обучением похоже связаны с мышечной слабостью, и в настоящее время нет достаточно информации, свидетельствующей о том, что мутации гена дистрофина влияют на познавательные способности.
У ребенка также может быть трудное поведение, которое влияет на общение мальчика дома, в школе и во всех сферах его жизни, и в этой области ему и его семье необходима будет поддержка и помощь.
Как осуществляется контроль ребенка с МДД?
Как уже было упомянуто выше, в настоящее время нет лечения, которое может справиться с прогрессирующей мышечной слабостью на длительный период. Однако, есть способы контроля, которые могут помочь облегчить состояние путем минимизирования вторичных осложнений разрушения мышц. Эти способы будут меняться по мере прогрессирования болезни. Группа специалистов-медиков поможет больным МДД и их семьям подготовить и осуществить эту программу контроля. В эту группу, возглавляемую детским неврологом, поначалу входят физиотерапевт и специалист по трудотерапии, а также специалисты в других областях по мере необходимости.
Упражнения
Как активные, так и пассивные упражнения являются важным аспектом контроля мальчиков с МДД. Мальчик в возрасте до восьми лет будет участвовать в нормальных игровых упражнениях насколько он способен. Важно не доводить его до точки изнеможения; слишком много упражнений могут дополнительно повредить мышцы. Плавание или гидротерапия являются одним из способов тренировки без лишнего напряжения на мышцы. Подъемная сила воды значительно помогает слабым мышцам, а удерживание дыхания под водой позволяет тренировать дыхательные мышцы. Упражнения этого типа могут легко выполняться даже по прошествии долгого времени после того, как ходить стало слишком тяжело. Некоторые мальчики продолжают с удовольствием заниматься водными видами спорта, даже когда им уже далеко за двадцать.
Приблизительно в шести до восьми лет и позднее мальчикам понадобится ручное кресло-коляска для перемещения на более длинные расстояния. Начиная приблизительно с девяти лет электрическое кресло-коляска или кресло-коляска с приводом позволит мальчикам сохранить свою независимость, когда ходьба становится слишком трудной, а падения слишком частыми. Многие мальчики продолжают заниматься спортом со своими ровесниками, пользуясь креслами-колясками. Некоторые, по мере взросления начинают заниматься организованным спортом на кресле-коляске.
Пассивные упражнения или вспомогательная растяжка должны быть разработаны как можно раньше. При участии физиотерапевта разрабатываются программы упражнений, целью которых является максимальное предотвращение укорачивания или «контрактуры» мышц, которое ограничивает движение в суставах. Для ежедневного выполнения этих упражнений требуется помощь родителей и/или ухаживающего персонала.
Поддерживающее оборудование
Чтобы обеспечить более длительную растяжку в икрах и лодыжках, назначаются ночные шины. Эти шины помогут поддержать голеностопный сустав в нормальном положении и предотвратить развитие контрактур (напряжения мышц). При постоянном пользовании креслом-коляской использование рамы для стояния в отдельных эпизодах школьного дня также может помочь. Стояние может улучшить общее кровообращение и обеспечить растяжку на уровне лодыжек, бедер и коленей. Системы для сидячего положения обеспечивают индивидуальную поддержку для сохранения прямого положения в кресле-коляске и для обеспечения комфорта. По мере того, как болезнь прогрессирует и мобильность снижается, ручное кресло-коляску будет необходимо заменить на кресло-коляску с приводом, которое поможет обеспечить максимальный уровень независимости. По мере возникновения необходимости будут использоваться другие типы поддерживающего оборудования и консультацию о них можно будет получить у физиотерапевта, специалиста по трудотерапии и в Ассоциации мышечной дистрофии.
Желательно рассмотреть оптимальность домашних условий не ранней стадии, с тем чтобы со временем вносить постепенные изменения. Например, когда понадобиться кресло-коляска с приводом, также потребуются подходящий автомобиль для перевозки, доступ к нему и прилегающее место парковки. Будет необходимо место для наклонного въезда, а двери должны быть достаточно широкими, чтобы сквозь них проходило кресло-коляска. Если в вашем доме есть ступеньки, это усложнит доступ. Обычно необходимо модифицировать ванную комнату, с тем чтобы можно было использовать оборудование для помощи при купании, одевании и туалетных процедурах.
Медицинское лечение
Доказано, что лекарственные препараты в форме лечения стероидами уменьшают потерю мышечного функционирования, и тем самым временно продляют мобильность мальчика. Тем не менее, сохранение способности ходить немного дольше не влияет на ожидаемую продолжительность жизни больного. Если невролог ребенка после обсуждения с ребенком и членами его семьи сочтет эту форму лечения пригодной, он решит, в каком возрасте это лечение должно начаться и какова должна быть программа этого лечения. Если данный вариант будет выбран, семью подробно проинформируют о возможных побочных эффектах. Наиболее значительными из них являются увеличение веса, остеопороз (пористость костей) и проблемы в поведении.
Питание
Чрезмерное увеличение веса, приводящее к ожирению, может возникнуть не только в качестве побочного эффекта лечения стероидами, но также из-за пониженной активности в результате ослабления мышц. Ожирение может отрицательно повлиять на функцию дыхания, работу сердца и кишечника (у больных МДД часто случаются запоры), а также создает трудности при подъеме ребенка членами семьи или ухаживающими лицами. Для предотвращения чрезмерного увеличения веса очень важно следить за ним и соблюдать хорошо сбалансированную диету. Родители, бабушки и дедушки, другие члены семьи и друзья могут помочь мальчику с МДД, если будут давать ему здоровую пищу с высоким содержанием клетчатки, свежих фруктов и овощей, а также побольше жидкости. Следует ограничить продукты с высоким содержанием жира и сахара.
Мальчики и подростки с чрезмерно большим или чрезмерно малым весом должны получать консультации у лечащего врача или диетолога с целью контроля питания и веса.
Также важно понимать, что потребление больше белка не окажет никакого воздействия на недостаток белка в мышцах мальчиков, больных МДД.
Хирургическое вмешательство
Иногда для устранения контрактуры (напряжения) на голеностопных суставах требуется хирургическое вмешательство. Обычно эта процедура выполняется, когда ребенку постоянно необходимо пользоваться креслом-коляской. Она помогает улучшить положение ступней для носки обуви, улучшает комфортность положения ступней в неподвижном состоянии и, в некоторых случаях, позволяет дольше сохранять мобильность. По мере того, как спинные мышцы становятся все слабее и мобильность снижается, позвоночник может начать изгибаться – это называется «сколиозом». Сколиоз, в свою очередь, приводит к другим осложнениям, таким как нарушение дыхания и постуральный дискомфорт при сидении и при сне. Рекомендуется исправлять сколиоз путем хирургического вмешательства в идеальный момент во время так называемого «окна возможности», при определении которого принимается в расчет этап взросления мальчика. Группа медиков, возглавляемая неврологом, обсудит этот вариант вмешательства с мальчиком, больным МДД, и членами его семьи за долго до того, как данная хирургическая операция станет необходима. Хирург-ортопед также выскажет свое мнение по этому поводу. Мальчики с болезнью Дюшенна, которым была сделана такая операция «слияния суставов позвоночника», обычно очень довольны результатом.
Дыхательная функция
По мере ослабления дыхательных мышц происходит постепенное ухудшение функционирования легких. Большое значение приобретают раннее лечение простудных заболеваний и профилактика инфекций грудной клетки. Больным МДД нельзя курить или находится в помещении, наполненном дымом от курения членов семьи или друзей. Операция с целью предотвращения сколиоза и специальные физиотерапевтические приемы могут помочь в поддержании функционирования легких. Однако, по мере снижения мобильности, когда ребенку постоянно необходимо пользоваться креслом-коляской, последующие нарушения дыхательной функции становятся практически неизбежными. Эти нарушения часто начинаются ночью и приводят к нарушению сна. Члены семьи и ухаживающие лица должны внимательно следить за появлением симптомов утренней вялости, отсутствия концентрации, головной боли; спутанности и сонливости днем, а также бессонницы ночью, когда у больных возрастает необходимость повернуться. Необходимы регулярные проверки специалистом по заболеваниям дыхательной системы.
Когда это становится необходимо, применяются неинвазивные аппараты для искусственной вентиляции легких, чтобы улучшить дыхание ночью и добиться эффективного сна. Больные МДД часто выбирают этот вариант, который помогает им поддерживать удовлетворительное качество жизни. В этом случае требуется особое внимание со стороны надежных ухаживающих лиц, будь то родители, если больной находится дома, или профессиональный ухаживающий персонал, если больной живет отдельно.
Образование
Следует рассмотреть два способа обучения: в обычной школе или специальное обучение. В Австралии все больные МДД дети имеют право посещать обычную школу. Для каждого ребенка должен быть составлен индивидуальный образовательный план (individualised education plan (IEP)) для получения надлежащей помощи в удовлетворении физических и образовательных потребностей ребенка. Сюда входит возможность получения компьютерных навыков. Они пригодятся ему позднее, когда из-за мышечной слабости станет трудно писать рукой. Необходимость использования компьютера будет возрастать в подростковом возрасте. Школа, где учится ваш ребенок, должна обратиться за получением дополнительных государственных фондов, называемых интеграционными фондами, которые обеспечат получение вашим сыном достаточной помощи (дополнительного времени с учителем) в школе для полного освоения учебного плана. Эти интеграционные фонды также позволяют школе пользоваться услугами физиотерапевта и специалиста по трудотерапии, которые будут регулярно проверять потребности ребенка и обеспечат соответствие школьных условий изменяющимся потребностям ребенка. Размер интеграционных фондов должен пересматриваться каждый год. Если у ребенка есть трудности с речью и общением, может поощряться развитие творческих навыков. Зачастую положение мальчиков с МДД еще больше осложняется отношением окружающих их лиц – их могут чересчур защищать или снисходительно к ним относится. Важно, чтобы таким детям были предложены образовательные возможности, которые позволят им быть продуктивными и создадут умственное стимулирование.
Паллиативная помощь
Трудности восприятия неизбежности смерти от МДД могут иногда помешать больному примириться с таким окончательным исходом. Непрерывное открытое общение с семьей имеет большое значение и отвечать на возникающие у мальчика вопросы следует честно, но с тактом. Семейный врач и врачи-специалисты являются важными членами группы поддержки на протяжении всей болезни и особенно на последней стадии. Ассоциация мышечной дистрофии (MDA) также может предоставить информацию и помочь семьям, которым нужна поддержка в примирении с горем и потерей.
Как мы можем помочь нашему сыну?
Открытое общение очень важно в процессе примирения. Если родители могут открыто и честно говорить друг с другом, делиться беспокойствами и обсуждать пути совладания с трудными ситуациями, это будет хорошо для всей семьи. Первым делом задайте медикам и другим членам персонала поддержки все имеющиеся у вас вопросы. Прочитайте все, что вы сможете найти об МДД, и регулярно читайте новые материалы о развитии в данной сфере. Ваш сын будет задавать вам вопросы, и вы будете лучше подготовлены к тому, чтобы ответить на них открыто и честно, если вы прочитали всю имеющуюся информацию. Слушайте его внимательно и обсуждайте задаваемые им вопросы и затрагиваемые проблемы. Будьте открытыми и простыми в общении. Поощряйте его говорить о его нуждах и учиться спрашивать других о помощи, когда она ему нужна, и вежливо отказываться, когда он сам может справиться.
Как мы уже отмечали, курение может быть особенно вредно для больного МДД, и вся семья должна знать об этом. Может быть рекомендована здоровая диета с ограничением пищевых продуктов с высоким содержанием жиров и сахара. Следует потреблять побольше жидкостей, фруктов и овощей. Будучи родителями, вы можете поощрять различные интересы и физические упражнения, такие как плавание, в соответствии с уровнем мобильности ребенка, а также дружбу с его ровесниками.
Не следует чрезмерно защищать своего сына. Ему нужна поддержка, любовь, теплое и заботливое семейное окружение и, не в последнюю очередь, поощрение жить насколько возможно полной жизнью с участием друзей и семьи. Многие семьи с больными МДД придумывают свои собственные виды деятельности и способны продолжать радоваться семейной жизни, а не ограничивать ее. Вам потребуется приспособиться к жизни с МДД, но эта болезнь не должна доминировать в вашей жизни.
Как быть с потребностями остальных членов семьи?
Рекомендуется вовлекать всех членов семьи, насколько это возможно, а не взваливать все дополнительные обязанности на одного человека. Каждому члену семьи нужна помощь, понимание и временная замена (передышка). Постарайтесь обеспечить баланс, чтобы было время и место для всех отношений – между родителями и другими детьми, между самими детьми и между родственниками и друзьями. Может быть полезно подружиться с другими семьями, в которых есть больные МДД или инвалиды.
Ассоциация мышечной дистрофия понимает, насколько тяжело родителям и другим членам семьи примириться с диагнозом МДД. Процесс примирения будет проходить через различные стадии. Будут моменты, когда вы будете испытывать чувства потери и горя, гнева, негодования и вины. Будут также моменты, когда вы будете испытывать довольно положительные эмоции и некоторую степень примирения. Консультанты отдела обслуживания клиентов Ассоциации мышечной дистрофии всегда готовы ответить на ваши вопросы и предоставить информацию и поддержку.
Если вам нужна дополнительная информация по любому из вопросов, затронутых выше, пожалуйста звоните в Ассоциацию мышечной дистрофии по телефону (03) 9320 9555 или посетите сайт The Home of MDA – www.mda.org.au.
Источник http://www.mda.org.au/Information/Translations/Russian/Russian.asp
ФОРМЫ МИОПАТИИ
ПРОГРЕССИРУЮЩАЯ МЫШЕЧНАЯ ДИСТРОФИЯ ДЮШЕННА |
||
Первые признаки |
От 1 до 5 лет |
|
Симптомы |
Обычно первыми поражаются мышцы тазового пояса и проксимальных отделов нижних конечностей. Характерны псевдогипертрофии икроножных, дельтовидных, четырехглавых и трехглавых мышц. |
|
Прогресс |
Болезнь прогрессирует быстро и приводит к летальному исходу до 25 летнего возраста. |
|
Тип наследования |
Х-сцепленный-рецессивный (женщины являются носителями) |
|
—————————-
ПРОГРЕССИРУЮЩАЯ МЫШЕЧНАЯ ДИСТРОФИЯ БЕКЕРА |
||
Первые признаки |
Проявляются обычно от 10 до 20 лет |
|
Симптомы |
Практически полностью идентичны Дюшенна, но имеют меньшую степень выраженности. Характерной особенностью этого типа ПМД является вовлечение в патологический процесс миокарда |
|
Прогресс |
Заболевание прогрессирует достаточно медленно и в большинстве случаев приводит к инвалидизации больного не ранее 40-летнего возраста |
|
Тип наследования |
Х-сцепленный-рецессивный (женщины являются носителями) |
|
—————————-
ПРОГРЕССИРУЮЩАЯ МЫШЕЧНАЯ ДИСТРОФИЯ ЭМЕРИ-ДРЕЙФУСА |
||
Первые признаки |
От 4 до 20 лет |
|
Симптомы |
Слабость мышц плечевого пояса и бицепсов. Часто встречаются контрактуры |
|
Прогресс |
Болезнь обычно прогрессирует медленно с вовлечением в процесс сердечной мышцы |
|
Тип наследования |
Чаще всего Х-сцепленный (женщины являются носителями). Реже аутосомно-доминантный и наиболее редко встречается аутосомно-рецессивный тип наследования |
|
—————————-
ПОЯСНОКОНЕЧНОСТНЫЕ ПРОГРЕССИРУЮЩИЕ МЫШЕЧНЫЕ ДИСТРОФИИ (мышечная дистрофия Эрба-Рота) |
||
Первые признаки |
Могут проявляться от 2 до 40 лет |
|
Симптомы |
Слабость мышц плечевого и тазовых поясов. «Утиная походка», гиперлордоз в поясничном отделе позвоночника, «крыловидные» лопатки, симптом дряблых надплечий |
|
Прогресс |
Течение заболевания умеренно прогрессирующее, приводящее к инвалидизации во взрослом возрасте |
|
Тип наследования |
Основное количество ПКМД (приблизительно 85%) наследуется аутосомно-рецессивно, 10% — аутосомно-доминантно и лишь 5% Х-сцепленно-рецессивно |
|
—————————-
ЛИЦЕ-ЛОПАТОЧНО-ПЛЕЧЕВАЯ ПРОГРЕССИРУЮЩАЯ МЫШЕЧНАЯ ДИСТРОФИЯ ЛАНДУЗИ-ДЕЖЕРИНА |
||
Первые признаки |
Обычно от 10 до 20 лет |
|
Симптомы |
Первыми поражаются мышцы лица, мышцы плечевого пояса |
|
Прогресс |
Прогрессирует медленно с некоторыми периодами быстрого ухудшения. Болезнь может длиться десятилетиями |
|
Тип наследования |
Аутосомно-доминантный |
|
—————————-
СПИНАЛЬНАЯ АМИОТРОФИЯ Верднига-Гофманна (1 ТИПА) |
||
Первые признаки |
С рождения до 6-ти месяцев |
|
Симптомы |
Первыми поражаются мышцы проксимальных отделов нижних конечностей и процесс имеет восходящее распространение |
|
Прогресс |
В целом продолжительность жизни ограничена 2 годами. Считается, что только 10%- 12% больных переживают пятилетний возраст |
|
Тип наследования |
Аутосомно-рецессивный |
|
—————————-
СПИНАЛЬНАЯ АМИОТРОФИЯ 2 ТИПА |
||
Первые признаки |
От 6-ти до 18-ти месяцев |
|
Симптомы |
Для этой формы заболевания характерны фасцикулярные подергивания кистей, языка, плечевого и тазового пояса, тремор кончиков пальцев вытянутых рук, контрактуры в суставах и деформации позвоночника |
|
Прогресс |
Обычно продолжительность жизни составляет 10-12 лет |
|
Тип наследования |
Аутосомно-рецессивный |
|
—————————-
СПИНАЛЬНАЯ АМИОТРОФИЯ Кугельберга-Веландер |
||
Первые признаки |
Чаще всего в возрасте 2-7 лет |
|
Симптомы |
Слабость в проксимальных группах мышц тазового пояса |
|
Прогресс |
Болезнь прогрессирует медленно. Инвалидная коляска обычно используется в более позднем периоде жизни. Продолжительность жизни не затронута |
|
Тип наследования |
Аутосомно-рецессивный |
|
—————————-
НАСЛЕДСТВЕННЫЕ МОТОРНО-СЕНСОРНЫЕ НЕЙРОПАТИИ |
||
Первые признаки |
Чаще всего заболевание дебютирует в 1-2 десятилетии жизни |
|
Симптомы |
Слабость и атрофия мышц дистальных отделов рук и ног, с последующей их деформацией |
|
Прогресс |
Болезнь прогрессирует медленно, но возможны индивидуальное течение. Продолжительность жизни нормальная |
|
Тип наследования |
В большинстве случаев аутосомно-доминантный |
|
Форма Роттауфа — Мортье — Бейера
Описана впервые в 1971 г. Ее можно охарактеризовать как фиброзирующую форму, поскольку ведущим симптомом является раннее развитие тяжелейших контрактур. Роттауф наблюдал 17 больных в 4 поколениях. Первые симптомы появлялись к концу первой декады жизни и выражались в развитии сухожильных ретракций и контрактур, вначале в виде ограничения тыльного сгибания стоп, затем сгибания шеи, разгибания в локтевых суставах.
Постепенно формируются патологические позы головы и туловища из-за развития фиброза мышц с невозможностью сгибания позвоночника. Очень медленно развиваются мышечные атрофии, мышечная слабость обычно выражена умеренно. Преобладают парезы и гипотрофии в лопаточно-плечевой области и в дистальных отделах ног. Полностью отсутствуют псевдогипертрофии. Интеллект сохранен, иногда даже выше нормы.
Прогрессирование заболевания медленное, к 40 годам больные могут быть еще подвижны, имеют нормальную фертильность. Исход заболевания в большинстве случаев летальный в связи с поражением сердечной мышцы и наличием атриовентрикулярного блока с соответствующими изменениями на ЭКГ.
ЭМГ и данные биопсии указывают на мышечный тип изменений. КФК и другие ферменты выявляют повышенную активность у молодых больных, в далеко зашедшей стадии она снижается. У гетерозиготных носителей нет клинических проявлений. Показатели активности ферментов у них нормальные.
Под нашим наблюдением находился больной Б., 27 лет, с подобной формой заболевания. Поступил в клинику нервных болезней с жалобами на резкое ограничение движений головы, особенно вперед и в стороны, а также в локтевых, коленных и голеностопных суставах, изменение походки, быструю утомляемость.
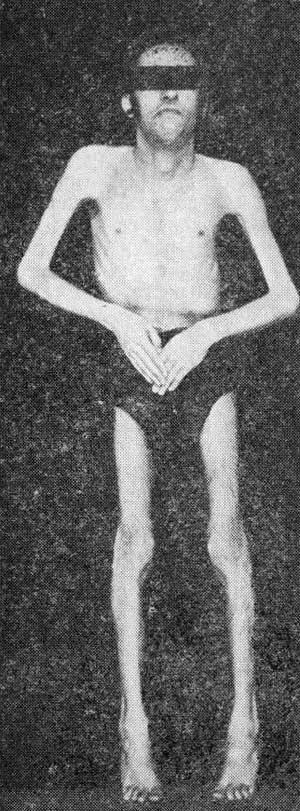
В семье аналогичных заболеваний не отмечается. Родился от 2-й беременности, протекавшей нормально, вовремя стал сидеть, ходить. Всегда отличался плохим аппетитом, был очень худым. В 8 — 9 лет отмечено изменение походки, стал ходить на пальцах, однако мог бегать, хорошо катался на велосипеде. В 11 — 12 лет появилось некоторое затруднения при сгибании головы. Очень медленно заболевание прогрессировало. К 16 — 17 годам развилась грубая деформация стоп с резко выраженными ретракциями ахилловых сухожилий.
Появилась деформация грудной клетки и S-образный сколиоз. Несмотря на это, значительного ограничения трудоспособности не было, хорошо учился в школе, поступил в университет, который успешно окончил, активно работает.
Значительное ухудшение состояния отмечает за последние 3 — 4 года, когда резко усилилась деформация позвоночника, нижних конечностей, появились грубо выраженные контрактуры в локтевых суставах. Стал быстро уставать, испытывает затруднения при ходьбе по лестнице, при вставании с низкого стула. На фоне общего похудания выявились четкие атрофии мышц верхних и нижних конечностей, больше в проксимальных отделах.
В неврологическом статусе: диффузная атрофия мышц конечностей (больше выраженная в проксимальных отделах), мышц плечевого и тазового поясов, мышц спины. Сила в руках и ногах в дистальных отделах до 5 баллов, в проксимальных — до 4 баллов. Резко снижена сила мышц передней поверхности шеи, мышц спины.
Встает с постели, прибегая к характерным вспомогательным приемам. Сухожильные рефлексы отсутствуют. Имеются грубейшие ретракции, особенно в мышцах задней поверхности шеи.
Контрактура в локтевых суставах составляет угол в 85°, значительно выражена ретракция ахилловых сухожилий. В связи с резко выраженной ретракцией мышц шеи создалась патологическая поза в виде запрокинутой головы назад, при ходьбе придерживает голову сзади рукой.
Контрактуры и ретракции доминируют в клинической картине над парезами и атрофиями мышц. Имеются изменения скелета в виде истончения диафизов длинных трубчатых костей, резкого искривления позвоночника, деформации грудной клетки с образованием реберного горба.
Обращает на себя внимание резкое снижение массы тела (40 кг при росте 162 см).
Ферменты сыворотки крови: Ф-1,6-Ф-альдолаза — 19,5 ЕД, КФК — 250 ME, СДГ — 0,6 ME, ACT — 66 ЕД, АЛТ — 116 ЕД, ЛДГ — 350 МЕ/мл. Креатин мочи — 0,39 г/сут, креатинин — 0,56 г/сут.
ЭКГ — частичное нарушение внутрижелудочковой проводимости по правой ножке пучка Гиса, синоаурикулярная блокада, экстрасистолия.
Рентгенограмма грудного и поясничного отделов позвоночника: выраженный S-образный сколиоз с ротацией по оси с образованием реберного горба справа. Биопсия дельтовидной мышцы справа выявила неравномерность окраски, разнокалиберность мышечных волокон. Атрофированные мышечные волокна имеют угловатую форму, расположены по 3 — 5 волокон вместе. Умеренное разрастание эндоперимизиальной соединительной ткани.
Заключение: в мышечном биоптате отмечается сочетание первично-мышечных и нейрогенных изменений. Анализы крови и мочи без патологии.
Консультация окулиста: острота зрения 0,2 с коррекцией +8,0 — 0,9. Цветоощущение не нарушено.
Дно глаза — диск зрительного нерва розовый, границы четкие, вены умеренно расширены. Таким образом, в нашем наблюдении симптоматика полностью укладывается в клиническую картину болезни Роттауфа. Случай относится к спорадическим. У 2 сестер больного детей не имеется, по линии матери нет родственников мужского пола.
«Нервно-мышечные болезни», Б.М.Гехт, Н.А.Ильина
ФАКТЫ О МЫШЕЧНОЙ ДИСТРОФИИ ДЮШЕННА/БЕККЕРА
По материалам Американской Ассоциации Мышечных дистрофий
ЧТО ТАКОЕ – МЫШЕЧНАЯ ДИСТРОФИЯ ДЮШЕННА/БЕККЕРА?
Мышечные дистрофии – это генетические заболевания, характеризующиеся прогрессирующим истощением и слабостью мышц, начинающихся с микроскопических изменений в них. По мере того, как мышцы разрушаются – их сила уменьшается.
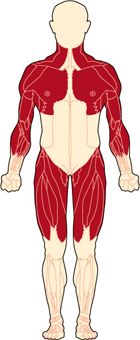
На ранних стадиях МДД и МДБ поражаются мышцы груди (сдвигающие плечи), туловища, а также верхние и нижние мышцы ног. Слабость этих мышц вызывает трудности во вставании, подъёме по ступенькам и удержании равновесия..
Мышечная дистрофия Дюшенна (МДД) впервые была описана в 1860 году французским неврологом Guillaume Benjamin Amand Duchenne. Мышечная дистрофия Беккера (МДБ) названа пo имени немецкого доктора Peter Emil Becker, описавшего этот вариант МДД в 1950 году.
При МДД признаки мышечной слабости обычно проявляются у мальчиков в возрасте около 3 лет. Болезнь постепенно ослабляет скелетные или произвольные мышцы рук, ног и туловища. Примерно в раннем подростковом возрасте или даже раньше могут быть затронуты также сердце и дыхательные мышцы.
МДБ – более мягкая форма МДД. Ее начало обычно приходится на подростковый возраст или раннюю юность, и протекает она медленнее и намного менее предсказуемо, чем МДД.
(Хотя МДД и МДБ болеют почти исключительно мальчики, в редких случаях этими заболеваниями могут болеть и девочки. См. «Это семейное?»)
ЧЕМ ВЫЗВАНЫ МЫШЕЧНЫЕ ДИСТРОФИИ ДЮШЕННА И БЕККЕРА?
До 1980 года было очень мало известно о причинах любых типов мышечных дистрофий. В 1986 году исследователи идентифицировали ген, дефекты в котором, называемые мутациями, вызывали МДД. В 1987 году ассоциированный с этим геном протеин был идентифицирован и назван дистрофином
Гены содержат в себе коды, или рецепты, для протеинов (белков), являющихся очень важными биологическими компонентами всех форм жизни. МДД развивается тогда, когда определенный ген, расположенный в Х-хромосоме, теряет способность вырабатывать протеин дистрофин. МДБ вызвана несколько иными мутациями в том же самом гене. У людей с МДБ присутствует некоторое количество дистрофина, однако его либо недостаточно, либо ухудшено его качество. Наличие некоторого количества дистрофина при МДБ предохраняет мышцы от столь же тяжелой и быстрой дегенерации, как при МДД.
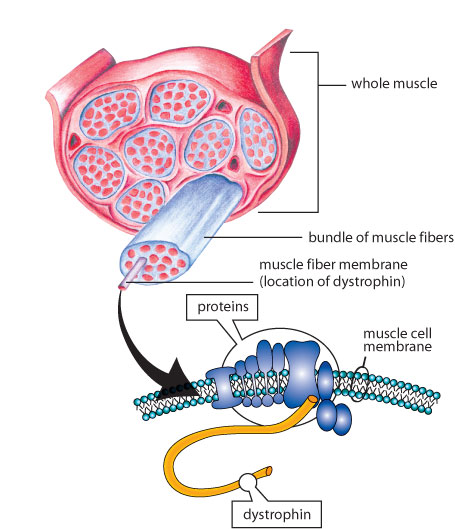
Мышцы состоят из пучков волокон (клеток). Группа независимых белков, расположенных вдоль мембраны, окружающей каждое волокно, помогают нормально функционировать мышечным клеткам. Когда один из этих белков, дистрофин, отсутствует – это вызывает мышечную дистрофию Дюшенна, если же его недостаточно, или он неполноценный – развивается мышечная дистрофия Беккера
Между прочим, употребление или неупотребление пищи, богатой протеинами, не может возместить утерянный дистрофин. Подробнее о том, как мутация гена приводит к развитию дистрофий Дюшенна и Беккера – см. «Это семейное?»
ЧТО ПРОИСХОДИТ С МЫШЦАМИ ЛЮДЕЙ С ДИСТРОФИЯМИ ДЮШЕННА И БЕККЕРА?
Миодистрофия Дюшенна
Развитие МДД довольно предсказуемо. Дети с этим заболеванием часто поздно начинают ходить. В этот период родители могут заметить увеличение икроножных мышц, или гипертрофию. В дошкольном возрасте дети с МДД могут выглядеть неловкими и часто падать. Вскоре появляются проблемы с подъёмом по лестнице, вставанием с пола или бегом.
В школьном возрасте дети могут начать ходить на пальцах или подушечках пальцев ног с немного перекатывающейся походкой. Походка становится переваливающейся и неустойчивой, и они легко могут споткнуться и упасть. Пытаясь удержать равновесие, они выпячивают живот и откидывают назад плечи. Также возникают трудности с подъёмом рук.
Почти все дети с МДД теряют способность к хождению в период от 7 до 12 лет. В юношеском возрасте для активности рук, ног и туловища требуется помощь или механические приспособления.
Миодистрофия Беккера
Часто диагноз «мышечная дистрофия Беккера» нельзя поставить до подросткового возраста или даже ранней юности, например, когда молодые люди замечают, что у них возникают трудности с занятиями физкультурой или военной подготовкой. Пытаясь скомпенсировать слабость мышц, мальчики начинают ходить переваливающейся походкой, на пальцах ног или подушечках пальцев, выпячивая живот.
Как и при МДД, истощение мышц при МДБ обычно начинается с бедренной и тазовой области, бедер и плеч. Однако при МДБ степень мышечной дегенерации отличается у разных людей в широких пределах. Одним требуется инвалидное кресло к возрасту 30 лет или чуть позже, в то время как другие долгие годы обходятся минимальными приспособлениями, такими, как трость.
КАКИЕ ТЕСТЫ ИСПОЛЬЗУЮТСЯ ДЛЯ ДИАГНОСТИКИ МДД/МДБ?
В диагностике любых форм мышечной дистрофии доктор обычно начинает со знакомства с пациентом и семейной историей и проведения медосмотра. Из этого можно почерпнуть довольно много, включая характер слабости. С истории и осмотра начинается долгий путь к предстоящему установлению диагноза еще до того, как будут проведены любые более сложные диагностические тесты.
 |
Поскольку ослаблены мышцы ног, мальчики с МДД применяют характерный способ вставания с пола, называемый приемом Говерса. Вначале они опираются на руки и колени, затем поднимают таз, а затем «шагают» руками вверх по ногам, чтобы поднять все тело. |
Важно получить правильный диагноз, поскольку у других заболеваний некоторые симптомы совпадают с таковыми при МДД/Б. МДБ зачастую может быть невыявлена, либо ошибочно диагностирована как конечно-поясная мышечная дистрофия (КПМД) или спинальная мышечная атрофия (СМА). По этой причине важно провести как генетическое исследование, так и мышечную биопсию до того, как вынести решение, что это именно МДБ.
Доктор также хочет определить, вызвана ли слабость мышц проблемами в них самих, либо в нервах, осуществляющих контроль этими мышцами. Нервы, контролирующие мышцы, или мотонейроны, исходящие из спинного и головного мозга и протягивающиеся ко всем мышцам, могут вызывать мышечную слабость, похожую на ту, что вызвана проблемами в мышцах, хотя в действительности это разные вещи.
Обычно природа слабости может быть уточнена при медосмотре. Иногда проводится специальное исследование, называемое электромиография или исследование нервной проводимости. В процессе этого исследования измеряется электрическая активность мышц и стимулируются нервы, чтобы увидеть, лежит ли проблема в мышцах или нервах.
На раннем этапе диагностики доктора часто назначают исследование крови для определения уровня КФК. КФК обозначает креатинфосфокиназа, энзим, просачивающийся из поврежденных мышц. Когда уровень КФК в крови повышен, это обычно означает, что мышцы разрушаются в результате какого-то патологического процесса, такого как мышечная дистрофия или воспаление. Следовательно, высокий уровень КФК наводит на мысль, что мышечная слабость вызвана патологическими процессами в самих мышцах, но не может точно указать, какое именно это может быть мышечное заболевание.
Чтобы определить, какое заболевание является причиной проблем, доктор может назначить мышечную биопсию, хирургическое удаление небольшого кусочка мышцы у пациента. Исследуя этот образец, доктор может много сказать о том, что же в действительности происходит с мышцами. Современные методики позволяют на основании биопсии отличить мышечные дистрофии от воспалительных и иных заболеваний, а так же отличать различные формы дистрофий
Другие тесты с использованием биоптатов могут дать информацию о том, какой протеин присутствует в мышечных клетках, и присутствует ли он в нормальном количестве и на своем ли месте. Это может помочь отличить МДД (дистрофин отсутствует) и МДБ (наличие некоторого количества неполноценного дистрофина). Также может быть назначен МР (магнитный резонанс). Это безболезненное сканирование позволяет доктору визуально определить, что происходит внутри ослабленной мышцы.
Доступность диагностических исследований ДНК, в которых используются либо клетки крови, либо мышечные клетки для получения точной генетической информации, быстро развивается. Вы можете уточнить у своего врача или генетического консультанта, какие тесты доступны. Поскольку многие мужчины с МДБ (и некоторые с МДД) становятся отцами, важно точно знать, какое именно наследственное заболевание у человека. Сестры людей с МДД или МДБ также могут пройти тест, чтобы выяснить, являются ли они носительницами заболевания, поскольку в этом случае у них могут быть дети с этим заболеванием.
ЭТО СЕМЕЙНОЕ?
Узнав о том, что у ребенка – генетическое заболевание, такое как МДД или МДБ, смущенные родители часто спрашивают: «Но в нашей семье этого не было, как же это может быть генетическим?»
МДД может быть характерной для семьи, даже если она есть только у одного члена семьи. Это обусловлено механизмом наследования генетических заболеваний.
И МДД, и МДБ наследуются по так называемому Х-сцепленному типу. Это означает, что ген, мутация в котором и вызывает заболевание, расположен на Х-хромосоме
Каждый ребенок мужского пола получает от матери Х хромосому, а от отца – Y хромосому, которая и делает его мальчиком. Дети женского пола получают две Х хромосомы, по одной от каждого родителя.
Каждый сын, рожденный женщиной с мутацией в гене дистрофина в одной из двух ее Х-хромосом, имеет 50% вероятность унаследовать поврежденный ген и иметь МДД или МДБ. Каждая дочь такой женщины имеет 50% вероятность унаследовать мутацию и стать носительницей. У носительниц обычно нет симптомов заболевания, но у них могут быть дети с мутацией или заболеванием.
Так как же в семье, в которой не было случаев МДД или МДБ, вдруг рождается сын с этим заболеванием?
Этому может быть два объяснения:
Генетическая мутация, приводящая к развитию МДД или МДБ, может присутствовать у женщин на протяжении нескольких поколений, и никто не будет об этом знать. Возможно, не рождались мальчики с заболеванием, или, если даже мальчик в ранних поколениях был болен, родственники могли не знать, что это была за болезнь.
Другое объяснение – это то, что ребенок с МДД или МДБ имеет новую генетическую мутацию, возникшую в процессе его внутриутробного развития. Как только у кого нибудь появляется генетическое заболевание, даже если мутация – спонтанная (новая) у этого человека, он может передать его своему потомству.
Мужчины с МДД или МДБ не могут передать поврежденный ген своим сыновьям, поскольку передают им Y-хромосому, а не Х. Но они безусловно могут передать его своим дочерям, поскольку каждая дочь получает от отца только Х-хромосому. Они будут носительницами, и у каждого их сына будет 50% вероятность того, что у них будет это заболевание, и так далее.
Хороший способ узнать больше о типе наследования в вашей семье – поговорить со своим врачом или геноконсультантом. Можно также посмотреть брошюру «Генетика и нервномышечные заболевания».
ЖЕНЩИНЫ И МДД
Почему у девочек не бывает МДД или МДБ? Когда девочка наследует поврежденный ген от матери, обычно она также получает «здоровый» ген дистрофина от отца, вырабатывающий достаточно протеина для того, чтобы предотвратить развитие заболевание. Мальчики, наследующие мутантный ген – болеют, поскольку у них отсутствует второй ген дистрофина, компенсирующий повреждение первого.
Тем не менее, хотя у девочек обычно МДД или МДБ не проявляется в полном объеме, некоторые женщины – носительницы поврежденного гена все же до некоторой степени больны. Небольшой процент женщин – носительниц мутации – так называемые «выраженные носители», и болезнь проявляется у них в мягкой форме.
У таких женщин дефицит дистрофина может проявляться слабостью мышц спины, рук и ног и их быстрой утомляемостью. У выраженных носителей также есть проблемы с сердцем, которые могут выражаться одышкой или неспособностью выполнять простые упражнения. Проблемы с сердцем, если их не лечить, могут быть довольно серьезными и даже опасными для жизни.
Всем женщинам – потенциальным носительницам МДД/МДБ было бы разумно пройти полное диагностические обследование для выяснения своего статуса. В дальнейшем, если подтвердится носительство, регулярная оценка силы и наблюдения за сердцем могут помочь справиться с симптомами, склонными к ухудшению.
ЧТО МОЖЕТ БЫТЬ СДЕЛАНО ДЛЯ ЛЕЧЕНИЯ МДД/МДБ?
Благодаря достижениям во многих областях медицины существуют очень хорошие терапевтические методы, способные помочь при всех проявлениях мышечных дистрофий Дюшенна и Беккера. Эти методы воздействия постоянно совершенствуются. Используя все доступные методы, пациенты могут продлить себе бодрость, активность и продолжительность жизни.
Контрактуры
Влияние болезни можно существенно минимизировать, сохраняя тело насколько возможно гибким, прямым и подвижным. Существует несколько способов добиться этого.
По мере разрушения мышц у больных с мышечной дистрофией часто развивается малоподвижность суставов, называемая контрактурами. Если ими не заниматься, они станут достаточно серьезно выраженными, вызывая дискомфорт и ограничивая подвижность и гибкость. Контрактуры могут затронуть колени, бедра, ступни, локти, запястья и пальцы.
Тем не менее, существует много способов минимизировать и отсрочить контрактуры. Упражнения на амплитуду движений, выполняемые регулярно, помогают замедлить контрактуры посредством предохранения сухожилий от преждевременного укорочения. Очень важно, чтобы физиотерапевт показал вам, как правильно выполняются эти упражнения.
Шины на руках и голенях также могут помочь сохранить конечности растянутыми и подвижными, задерживая начало развития контрактур.
Когда контрактуры сформируются, ослабить их может помочь хирургическое вмешательство. Процедура удлинения сухожилия, называемая операцией на ахилловом сухожилии, часто проводится для лечения контрактур в щиколотке, когда ребенок еще сохраняет способность к хождению. Обычно ребенку требуется после этого использование шин на ногах.
Искривления позвоночника
У подростков с МДД позвоночник может постепенно принять искривленную форму. Это искривление может произойти из стороны в сторону (сколиоз), либо в продольном направлении с принятием формы горба (кифоз). Иногда у тех, кто еще ходит, наблюдается вогнутое искривление в поясничном отделе позвоночника, называемое лордозом.
Сильный сколиоз может мешать сидению, сну и даже дыханию, поэтому желательно его не допускать.
О том, какие упражнения необходимы для сохранения ровной спины, насколько это возможно, а также о правильных позициях во время сидения и сна, можно проконсультироваться с физиотерапевтом
Хирургический метод исправления искривления заключается во введении в позвоночник металлических стержней. Обычно такие операции проводятся в возрасте 11-13 лет
Медикаменты
Медикаментами являются группа препаратов, известных как кортикостероиды, продемонстрировавших эффективность в замедлении прогрессирования МДД (данных «за» или «против» кортикостероидов при МДБ недостаточно)
В 2005 году American Academy of Neurology опубликовала рекомендации по применению этих препаратов при МДД. Они заключаются в следующем:
•Преднизолон или дефлазакорт имеют эффект в терапии МДД. Семилетние исследования показали, что их применение увеличивает силу и улучшает временнЫе характеристики мышц (таких как например время, затраченное на подъем по ступенькам) а также функцию легких
•Эффективные начальные дозы: 0,75 мг на кг массы тела ежедневно для преднизолона, и 0,9 мг на кг массы тела – для дефлазакорта
•Доза должна быть снижена при наличии серьезных побочных эффектов, таких как значительное увеличение массы тела, истончение костей (остеопороз), а также поведенческие проблемы. Наиболее частые побочные эффекты – увеличение массы тела и формирование округлого одутловатого лица
•Пока нет четкой уверенности, что применение дефлазакорта имеет меньшие побочные эффекты, чем преднизолона
Оптимальный возраст начала кортикостероидной терапии не определен. Некоторые врачи уверены, что ее необходимо начинать сразу после постановки диагноза, в то врема как другие предпочитают дождаться времени, когда у мальчиков возникают первые проблемы с хождением. До начала кортикостероидной терапии врач должен обсудить с родителями ожидаемые положительные и потенциальные побочные эффекты
В сочетании с преднизолоном часто назначаются кальциевые добавки и витамин D для нейтрализации его нежелательного воздействия на кости
Обычно рекомендуется низкокалорийная диета с малым содержанием натрия для регулирования массы тела и наблюдаемой при приеме кортикостероидов задержке жидкости.
Иногда при МДД или МДБ назначаются препараты для снижения нагрузки на сердце (см. «Как еще МДД и МДБ воздействуют на организм?»)
Фиксаторы, вертикализаторы и кресла-коляски
Фиксаторы, также называемые ортезами, поддерживают голень и стопу, или охватывают колено.
Голеностопные ортезы иногда назначают для применения ночью, чтобы предотвратить отвисание стопы ребенка во время сна
Стояние в течение некоторого времени в течение дня, даже с минимальной весовой нагрузкой, содействует лучшей циркуляции, укреплению костей и выпрямлению позвоночника. Помочь в этом людям с МДД и МДБ могут ходунки или вертикализаторы. Некоторые кресла-коляски также имеют вертикальное положение
Рано или поздно всем мальчикам с МДД требуется кресло-коляска. Многие используют вначале кресла-коляски в школе или на прогулке, продолжая ходить дома. При МДД необходимость постоянного использования коляски наступает как правило в возрасте около 12 лет. Хотя многие дети и их родители воспринимают кресла-коляски как символ инвалидности, большинство считает, что их использование позволяет быть более мобильным, активным и независимым, нежели при попытках ходить во что бы то ни стало на очень слабых ногах.
Другие приспособления могут помочь тем, кто ухаживает за людьми с МДД или МДБ. Среди самых простых – пересадочные площадки для помощи в пересадке с коляски или на коляску. Также можно использовать механические (чаще – гидравлические) подъемники, складные стулья и кровати с электронным управлением.
КАК ЕЩЕ МДД И МДБ ВОЗДЕЙСТВУЮТ НА ОРГАНИЗМ?
Боль и чувствительность
Вы можете быть спокойны, зная, что истощение мышц при МДД и МДБ обычно безболезненно само по себе. Некоторые люди говорят о периодических мышечных спазмах, которые обычно снимаются безрецептурными болеутоляющими средствами
Также, поскольку мышечная дистрофия не затрагивает напрямую нервы, люди с этим заболеванием сохраняют нормальное осязание и другие чувства. Они также как правило контролируют гладкие, или непроизвольные мышцы мочевого пузыря и кишечника, и сохраняют нормальную сексуальную функцию.
Сердце
Аналогично мышцам конечностей, сердечная мышца также может быть ослаблена вследствие недостатка дистрофина. С течением времени, иногда еще до достижения возраста 10 лет, кардиологические проблемы в связи с МДД могут стать жизнеугрожающими. Таким образом, неоходимо тщательное наблюдение за сердечно-сосудистой системой, осуществляемое как правило детским кардиологом.
Вследствие дефицита дистрофина у людей с МДД и МДБ часто развивается кардиомиопатия – слабость сердечной мышцы. Мышечный слой сердца (миокард) вырождается так же, как и скелетные мышцы, что может привести к фатальным кардиологическим проблемам
У некоторых людей с МДБ не так серьезно выражено поражение скелетных мышц, но при этом – серьезные кардиологические проблемы
В 2005 American Academy of Pediatrics сформулировала рекомендации для людей с МДД и МДБ, а также носителей этих заболеваний.
Для пациентов с МДД рекомендовано проходить полное кардиологическое обследование в раннем детстве, и в дальнейшем – раз в 2 года до достижения возраста 10 лет. В дальнейшем обследование необходимо проводить каждый год, или при появлении симптомов слабости сердца, таких как задержка жидкости и одышка.
Для пациентов с МДБ рекомендовано обследование по крайней мере раз в два года, начиная с возраста 10 лет
У носителей МДД и МДБ риск развития кардиомиопатии выше среднего. Специалисты полагают, что носителям необходимо проходить полное кардиологические обследование в поздней юности или ранней зрелости, или скорее – когда проявляются симптомы, и в дальнейшем – проходить такие обследования каждые 5 лет, начиная с 25-30 летнего возраста
Есть предварительные данные, что лечение с применением ангиотензин-превращающих энзимов (АПФ) и бета-блокаторов может замедлить повреждение сердечной мышцы при МДД и МДБ, если лечение начать, как только обнаруживаются отклонения на эхокардиограмме (ультразвуковое исследование сердца), не дожидаясь появления симптомов
Некоторые пациенты с МДБ с серьезными кардиологическими проблемами на фоне неплохого общего состояния были успешно пролечены методом трансплантации сердца.
Дыхательная функция
Когда мальчики с МДД достигают возраста около 10 лет, диафрагма и другие мышцы, управляющие работой легких, ослабевают, и легкие уже менее эффективно выполняют свою функцию. Проблемы, сигнализирующие о недостаточной дыхательной функции – это головные боли, ослабление мыслительной активности, трудности с концентрацией или сохранением бодрости, кошмарные сновидения.
Люди с ослабленной дыхательной системой также более подвержены инфекциям и испытывают трудности при кашле. Простое переохлаждение может привести к развитию пневмонии. При развитии инфекции очень важно получить немедленную медицинскую помощь, дабы не допустить тяжелой дыхательной недостаточности
Когда дыхательная функция ослабевает, семья может приобрести аппарат вентиляции легких, либо научиться процедурам, способствующим откашливанию и сохранению бронхов свободными от выделений. Получить необходимую информацию можно у терапевта или пульмонолога
В некоторых случаях может потребоваться принудительная вентиляция для обеспечения достаточного движения воздуха в легкие и из легких. Иногда дыхательная маска требуется только ночью. Если это необходимо более часто, возможно проведение трахеотомии (для обеспечения поступления воздуха в легкие непосредственно в трахею вставляется трубка)
Существуют эффективные неинвазивные системы вентиляция, позволяющие не использовать трубки. Даже тем, у кого установлены трубки в трахее, иногда возможно отсоединять их от аппарата на некоторое время в течение дня. Более современные трубки имеют в конструкции клапаны, позволяющие говорить.
Интеллектуальные способности
Около трети мальчиков с МДД имеют некоторую степень отставания познавательных способностей, а некоторые – весьма серьезное. Специалисты полагают, что дефицит дистрофина в головном мозге может вызвать познавательные и поведенческие отклонения. Проблемы обучения, наблюдаемые у некоторых людей с МДД и МДБ, проявляются в трех основных областях: фокусировка внимания, словесное обучение и память, и эмоциональное взаимодействие.
Если есть подозрение, что у ребенка некоторая задержка в умственном развитии, необходимо обратиться к детскому нейропсихологу. Если это подтверждается, необходимо незамедлительно начинать обучающее и психологическое вмешательство. Специалист может порекомендовать упражнения и другие способы для взаимодействия с ребенком, которые помогут ему восполнить этот дефицит.
МОГУТ ЛИ СПЕЦИАЛЬНЫЕ ДИЕТЫ ИЛИ УПРАЖНЕНИЯ ПОМОЧЬ ПРИ МДД И МДБ?
Диета
Многие люди, услышав слова «утрата белка» логично задают вопрос: «Мне нужно потреблять больше белка?» К сожалению, потребление пищи с большим содержанием белка не оказывает никакого эффекта на белки, отсутствующие при мышечной дистрофии
Неизвестно ни о каких специальных диетических ограничениях или дополнениях, способных помочь при МДД или МДБ. Комбинация малоподвижности и слабости мышц живота может вызывать тяжелые запоры, поэтому диета должна быть с высоким содержанием жидкости и клетчатки, с преобладанием свежих фруктов и овощей.
У мальчиков, пользующихся электрическими колясками, принимающими преднизолон и не слишком активных вероятно необходимо ограничивать калорийность пищи для поддержания веса. Ожирение дает дополнительную нагрузку на и без того ослабленные мышцы и сердце. Специалисты считают, что низкокалорийная диета не оказывает никакого вредного воздействия на мышцы.
Для принимающих преднизолон и имеющих проблемы с сердцем также может понадобиться диета с низким содержанием натрия.
Упражнения
Упражнения могут помочь в формировании мышц, поддержании здоровой сердечнососудистой системы и способствовать хорошему самочувствию. Однако при мышечной дистрофии излишние упражнения могут повредить мышцы. Проконсультируйтесь с врачом о наилучшей физической нагрузке. При МДД и МДБ возможны умеренные физические нагрузки, но не до изнеможения.
Некоторые эксперты рекомендуют плавание и упражнения в воде (акватерапию) как хороший способ подержания тонуса мышц, насколько это возможно, не вызывая их избыточного перенапряжения.
Поддерживающая способность воды может помочь предупредить некоторые виды напряжения и повреждения мышц. До начала любой программы физических упражнений необходимо пройти кардиологическое обследование.
Физио- и трудотерапия
Физиотерапия обычно является частью комплексной терапии при МДД и МДБ. Для оценки физического состояния и разработки программы физиотерапии необходимо обратиться к физиотерапевту. Основные цели физиотерапии – сохранение подвижности суставов, предупреждение контрактур и сколиоза.
Трудотерапия более сфокусирована на специфической активности и функциях, в отличие от физиотерапии, акцентированной на мобильности, и, где возможно, усилении больших мышечных групп.
Трудотерапия может помочь в решении задач, связанных с работой, отдыхом и повседневной жизнью, таких как передвижение, одевание или пользование компьютером.
КАК С ЭТИМ ЖИТЬ?
Когда у одного из членов семьи МДД или МДБ, вся семья испытывает потребность в поддержке и эмоциональные реакции. Многие черпают помощь и поддержку из религиозных источников, общения с семьями с аналогичным опытом, книг по психологии или консультаций со специалистами. Такие специалисты обычно рекомендуют следующее:
Для детей
•Отвечайте на вопросы детей о болезни по мере их взросления честно и доступным языком
•Всегда представляйте ребенка как личность, с болезнью лишь как одним из аспектов его жизни
•Акцентируйтесь на том, что ребенок может сделать, и помогайте ему делать то, что он хочет. Дети часто находят способы для занятий спортом или другими увлечениями
•Воспитывайте его, как любого другого ребенка, запасаясь терпением, ответственностью, надеждой и любовью. Избегайте избыточной его опеки и помогайте стать независимым
•Предпринимайте нормальную семейную активность, включая отпуска и развлечения. Проявив воображение и терпение, вы можете найти способы делать почти все
Для семьи
•Будьте внимательны к эмоциям и уровню стресса друг друга, проявляйте терпение и доброжелательность
•Планируйте регулярный отдых от обязанностей по заботе
•Решайте проблемы, связанные с болезнью, по мере их поступления. Не фокусируйтесь на будущих осложнениях
•Дайте себе кредит по затрачиваемым усилиям и трудности своих обязанностей
•Организуйте команду поддержки и просите о помощи, когда это необходимо
Научные исследования для поиска методов лечения МДД
С 1986 года, когда был идентифицирован ген, мутации в котором вызывают МДД и МДБ, ученые далеко продвинулись в понимании механизмов этих заболеваний. В настоящее время разрабатываются несколько направлений в поиске способов того, как остановить или обратить разрушение мышц при этих заболеваниях
Одни исследователи создали работающий ген дистрофина без мутаций, и в настоящее время тестируют его безопасность в небольшом клиническом исследовании с участием мальчиков с МДД
Другие исследователи тестируют PTC124, препарат, изменяющий способ «чтения» клетками генетической информации. Примерно у 15% пациентов с МДД молекулярный стоп-сигнал расположен слишком рано для того, чтобы мог синтезироваться полноценный дистрофин. PTC124 заставляет клетки игнорировать этот сигнал
Еще одни исследователи экспериментируют с антисмысловыми нуклеотидами, составами, разработанными, чтобы побудить клетки обойти некоторые типы генетических ошибок, а не только стоп-сигнал. Эти составы прошли лабораторные тесты, и первые клинические испытания продемонстрировали многообещающие результаты
Другие группы исследователей используют стволовые клетки, выделенные из мышц, кровеносных сосудов или костного мозга в попытках добиться регенерации мышц
И, наконец, несколько групп разрабатывают стратегии усиления синтеза протеина атрофин, который очень похож на дистрофин, но синтезируется у людей с МДД и МДБ. Эксперименты показывают, что повышение уровней атрофина может до некоторой степени компенсировать дефицит дистрофина
Краткий обзор основных исследований можно прочесть здесь
Образовательная программа по неврологии: Миопатии
Лекцию читает д.м.н. проф. Ольга Петровна Сидорова
Образовательная программа по неврологии: Миопатия Ландузи-Дежерина
Лекцию читает д.м.н. Алексей Сергеевич Котов
Образовательная программа по неврологии: Болезнь Шарко-Мари
Лекцию читает д.м.н. Алексей Сергеевич Котов