

В основе врожденных полимиопатии лежат аномалии строения мышц, возникающие в эмбриональном периоде. Заболевания диагностируются в младенческом и раннем детском возрасте. Аномалии строения выявляются при гистохимическом исследовании замороженных срезов биоптатов мышц и при электронной микроскопии.
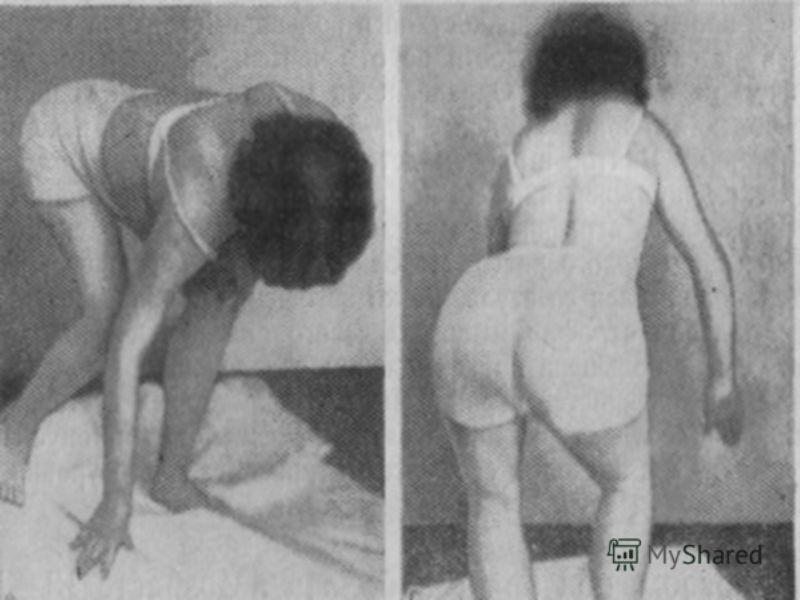
В большинстве случаев у больных младенцев обнаруживаются снижение мышечной силы, гипотония (снижение сопротивления при пассивных движениях в конечностях) и задержка двигательного развития. По мере роста ребенка наблюдается некоторое клиническое улучшение, но всегда сохраняются признаки мышечной патологии.
Во многих случаях в пожилом возрасте по неизвестной причине наблюдается незначительное нарастание двигательных расстройств. Дегенерация мышечных волокон при биопсии не обнаруживается, уровень КФК в крови обычно в пределах нормы, однако при ЭМГ часто выявляются миопатические изменения.
Другими представителями этой группы врожденных миопатии с относительно непрогрессирующим течением являются: миопатия центрального стержня, немалиновая миопатия, митохондриальные миопатии (с «рваными красными волокнами») и еще более редкие типы миопатии (саркотубулярные миопатии, миопатии с «зебровидными» тельцами, редуцированными тельцами, тельцами по типу отпечатков пальцев). При всех этих заболеваниях мышечная слабость более выражена в проксимальных отделах туловища и конечностей; поражение мышц глаз наблюдается при миопатии центрального стержня и при митохондриальных миопатиях. Дифференциально-диагностические признаки врожденных миопатии, многие из которых до сих пор спорны, в этой статье не рассматриваются. Они подробно обсуждаются Fardeau и Tome, Franzini-Armstrong, а также в работе Emery.
Спинальные амиотрофии.
О дегенеративных заболеваниях мотонейронов переднего рога уже упоминалось при описании заболеваний двигательной системы. Более подробно эти заболевания изложены в данном разделе в связи с тем, что с ними и с уже описанными ранее полимиопатиями проводится дифференциальный диагноз при синдроме вялого ребенка.
Первые проявления врожденной (тип наследования, как правило, аутосомно-рецессивный) дегенерации мотонейронов передних рогов спинного мозга могут обнаруживаться при рождении (мышечная гипотония, в некоторых случаях артрогрипоз) или в первые два месяца жизни. Летальный исход может наступить в результате дыхательной недостаточности. Медленно прогрессирующие формы спинальных амиотрофий дебютируют в позднем младенческом, раннем или даже позднем детском возрасте. Прогрессирующее нарушение функции мышц туловища, конечностей и лицевых мышц (за исключением мышц глаз) мешает двигательному развитию. Сухожильные рефлексы отсутствуют, но чувствительность сохранена, когнитивные функции не нарушены.
При наиболее распространенной аутосомно-рецессивной форме спинальной амиоатрофии с ранним дебютом (болезнь Верднига—Гоффмана) продолжительность жизни редко превышает 2—3 года, что связано с поражением бульбарной группы мышц, приводящим к дисфагии, дефициту питания и дыхательной недостаточности. Как правило, больные дети никогда не начинают сидеть, стоять и ходить. ЭМГ выявляет потенциалы фибрилляции и уменьшение количества двигательных единиц. Уровень креатининфосфокиназы в сыворотке не повышен, при биопсии мышц обнаруживается пучковая атрофия обоих типов мышечных волокон. Некоторые пациенты с дебютом заболевания в позднем младенческом или раннем детском возрасте доживают до подросткового или молодого возраста.
Наиболее легкая форма врожденной спинальной амиотрофии с дебютом в возрасте от 2 до 17 лет, при которой способность к самостоятельной ходьбе сохраняется у взрослых пациентов, была описана Вольфарт (Wohlfart) и соавт., а также Кугельбергом и Веландер. В этих случаях и иногда даже при более позднем дебюте страдают преимущественно мышцы проксимальных отделов конечностей или лицевые мышцы (синдром Фацио— Лонде). Симптомы другой формы прогрессирующей бульбарной и спинальной амиотрофии проявляются на четвертом-пятом десятилетии (синдром Кеннеди); тип наследования — рецессивный, сцепленный с полом; поражение мышц сочетается с атрофией яичек и низким уровнем андрогенов.
Лечение поддерживающее, симптоматическое.
По теме:
Спинальная амиотрофия
Общее понятие патологии
Амиотрофия спинальная детская Верднига-Гоффманна относится к группе заболеваний, при которых поражаются двигательные ядра, находящиеся в передних рогах спинного мозга. Это индуцирует процесс, в ходе которого происходит постепенная атрофия мышечной ткани. Заболевание начинает проявляться в раннем детстве (как правило, с трехмесячного возраста) и заканчивается развитием синдрома вялого ребенка. В течение нескольких лет наступает смерть.
Частота встречаемости: на сто тысяч новорожденных приходится всего лишь семь детей, у которых имеются симптомы данного заболевания.
Причины возникновения спинальной амиотрофии
Большинство вариантов спинальной амиотрофии вызвано мутациями, которые происходят в пятой хромосоме и наследуются по аутосомно-рецессивному типу. Изредка могут встречаться аутосомно-доминантный, Х-сцепленный рецессивный варианты наследования. Иногда возникают спорадические варианты.
Патоморфология
В передних рогах спинного мозга обнаруживаются недоразвитые клетки. Передние корешки подвергаются демиелинизации. Похожие изменения затрагивают ядра и корешки нижеперечисленных черепно-мозговых нервов:
- тройничный (V);
- отводящий (VI);
- лицевой (VII);
- языкоглоточный (IX);
- блуждающий (X);
- добавочный (XI);
- подъязычный (XII).
Для изменений скелетной мускулатуры характерно: пучковая атрофия, гиалиноз, гиперплазия соединительной ткани, гипертрофические процессы в отдельных мышечных волокнах.
Клиническая картина спинальной амиотрофии
Существует три формы амиотрофии спинальной детской Верднига-Гоффмана, различающиеся по времени, когда появляются первые симптомы, и по динамике развития миодистрофического процесса:
- врожденная;
- ранняя детская;
- поздняя.
Характеристика врожденной формы
 Вялые парезы возникают сразу же после рождения. Особенно часто наблюдаются мышечная гипертония, характеризующаяся генерализацией процесса, и отсутствие рефлексов с сухожилий. Быстро появляются бульбарные расстройства:
Вялые парезы возникают сразу же после рождения. Особенно часто наблюдаются мышечная гипертония, характеризующаяся генерализацией процесса, и отсутствие рефлексов с сухожилий. Быстро появляются бульбарные расстройства:
- вялое сосание;
- фибрилляции языка;
- слабый крик;
- снижение глоточного рефлекса.
Параллельно данная форма болезни Верднига-Гоффманна может сочетаться с различными костно-мышечными деформациями: сколиоз; контрактуры суставов; воронкообразная грудная клетка. Статические и локомоторные функции развиваются чрезвычайно медленно. Интеллект у таких детей обычно снижен. Нередко могут наблюдаться врожденные пороки развития:
- крипторхизм;
- косолапость;
- гемангиома;
- гидроцефалия;
- дисплазия тазобедренных суставов.
Течение врожденной формы: характеризуется злокачественностью и быстрым прогрессированием. Причины смерти – сердечнососудистая и дыхательная недостаточность – обусловлены слабостью грудных мышц.
Характеристика ранней детской формы
 Во второй половине первого года жизни проявляются первые признаки спинальной амиотрофии. Моторное развитие на первых трех месяцах существования – удовлетворительное. Дети начинают держать голову, сидеть, изредка ходить. Течение данной формы – подострое: она возникает, как правило, после перенесенной кишечной инфекции. Первоначальная локализация вялых парезов – ноги, затем – руки и туловище. Диффузная атрофия мышечной ткани в данном случае сочетается с:
Во второй половине первого года жизни проявляются первые признаки спинальной амиотрофии. Моторное развитие на первых трех месяцах существования – удовлетворительное. Дети начинают держать голову, сидеть, изредка ходить. Течение данной формы – подострое: она возникает, как правило, после перенесенной кишечной инфекции. Первоначальная локализация вялых парезов – ноги, затем – руки и туловище. Диффузная атрофия мышечной ткани в данном случае сочетается с:
- фасцикуляциями;
- тремором пальцев;
- языка;
- сухожильными контрактурами.
На поздних стадиях наблюдается бульбарный паралич, мышечная гипотония.
Течение ранней детской формы: протекает намного легче, по сравнению с врожденной формой. Смерть наступает обычно в возрасте до пятнадцати лет.
Характеристика поздней формы
Начинается в возрасте двух лет. К этому времени статические и локомоторные функции, как правило, развиты. Дети ходят, бегают. Начало процесса атрофии постепенное, медленное, практически незаметное: появляются неловкость и неуверенность в движениях, походка, характерная для «заводной куклы» – дети идут, при этом производя сгибание ног в коленях. К признакам поздней формы амиотрофии спинальной детской Верднига-Гоффманна относятся:
- вялые парезы с локализацией процесса в нижних конечностях (проксимальные группы мышц);
- малозаметность атрофического процесса вследствие хорошего развития подкожного слоя жировой клетчатки; снижение глоточного и небного рефлекса; угасание сухожильных и надкостничных рефлексов;
- костно-суставные деформации грудной клетки как проявления сочетанной патологии; тремор пальцев (мелкий);
- признаки бульбарного паралича;
Течение поздней формы: мягче, чем две вышеописанные формы. Больные живут до тридцати лет.
Диагностика болезни Верднига-Гоффманна
В основе постановки лежат следующие методы обследования: генеалогический анализ; лабораторная диагностика генетических аномалий; электромиография; получение биоптатов мышечной ткани с их последующим патоморфологическим исследованием; определение активности КФК; Особое внимание при этом уделяется особенностям клинической симптоматики:
- раннее начало амиотрофии спинальной детской Верднига-Гоффманна;
- локализация диффузных атрофий в проксимальных группах мышц;
- генерализованная мышечная гипотония;
- фасцикуляции и фибрилляции языка;
- отсутствие псевдогипертрофии;
- злокачественное течение.
 Дифференциальный диагноз:
Дифференциальный диагноз:
- при постановке диагноза амиотрофии Верднига-Гоффмана необходимо исключить;
- амиотонию Оппенгейма; мышечную дистрофию Дюшена и Эрба – Рота;
- детский церебральный паралич (атоническая форма);
- спинальную амиотрофию Кугельберга – Веландер;
- наследственные болезни обмена веществ;
- спондилез шейного отдела позвоночника;
- отравление свинцом.
Используемые методы лечения
Какие-либо эффективные методы лечения, способные воздействовать на причину амиотрофии спинальной детской Верднига-Гоффманна, отсутствуют. Используют препараты, которые улучшают трофические процессы нервной ткани. К ним относятся: церебролизин; аминолон; энцефабол. Таким пациентам назначают массаж и ЛФК.
Источник: pediatriya.info


