С.К. Евтушенко, М.Р. Шаймурзин, О.С. Евтушенко, Л.Ф. Евтушенко, Е.В. Дегонская, И.С. Евтушенко, Д.А. Сохань, Донецкий областной детский клинический центр нейрореабилитации, Донецкий национальный медицинский университет им. М. Горького
Резюме
В статье описаны этиология, патогенез, клиника, диагностика, дифференциальная диагностика, ЭМГ-характеристика и новые подходы к лечению наиболее часто встречающихся в практике детского невролога форм нейромышечных заболеваний. Дана новая, адаптированная для практического здравоохранения, международная классификация нейромышечных заболеваний. Представлены модифицированные стандарты диагностики и лечения детей с нейромышечной патологией, которые включают патогенетическую, специальную медикаментозную и немедикаментозную терапию с учетом степени тяжести, стадии заболевания, электромиографических данных. Предложены альтернативные методы лечения злокачественных форм мышечных дистрофий.
Ключевые слова
классификация, этиология, патогенез, диагностика, лечение нейромышечных заболеваний.
Нейромышечные заболевания (НМЗ) — клинически и генетически гетерогенная группа наследственно-дегенеративных заболеваний, характеризующаяся прогрессирующей мышечной слабостью и атрофиями. По данным В.Ю. Мартынюк (2005), в Украине было зарегистрировано 1540 человек, страдающих нейромышечной патологией. Ежегодно выявляется около 200 первичных больных, но, к сожалению, и смертность среди этой категории остается высокой.
На первом десятилетии жизни дебютирует большинство клинических форм прогрессирующих мышечных дистрофий (ПМД) и амиотрофий, имеющих прогредиентное течение и на сегодняшний день неизлечимых. Вместе с тем в последнее десятилетие достигнуты значительные успехи в изучении молекулярных механизмов наследственных нейромышечных заболеваний. Практически при всех формах нейромышечной патологии выявлены конкретные мутантные гены на хромосомах. При многих формах установлен продукт гена — белок, контролирующий мутантный ген, являющийся причиной первичного биохимического дефекта [7]. На основании мировых данных, новейших разработок в области молекулярной диагностики, превентивной диагностики и генной терапии в 9-м номере журнала «Neuromuscular Disorders» за 1999 год опубликована Клинико-молекулярная генетическая классификация мышечных дистрофий [26]. Согласно данной классификации, в настоящее время можно выделить дистрофинопатии, эмеринопатии, ламинопатии, конечностно-поясные мышечные дистрофии (МД), дистальные типы, окулофарингеальные типы, врожденные МД [7, 26].
Учитывая основы данной классификации и предыдущую классификацию нейромышечных заболеваний (ВОЗ, 1988), а также собственные данные по разработке методов ранней клинико-инструментальной диагностики, мы адаптировали Клинико-молекулярную классификацию за 1999 год в целях использования ее в клинической практике. Публикуя международную классификацию, мы прежде всего преследовали главную цель — представить и ознакомить детских неврологов с достаточно большим количеством форм нейромышечных заболеваний, их фенокопий, обширной группой структурных, метаболических миопатий. Ранний современный паттерн диагностики и лечебных мероприятий позволяет сегодня значительно улучшить качество жизни больных. После названия формы нейромышечного заболевания мы указали его международный номер (MIM), который соответствует составленному В. Мак-Кьюсиком каталогу генов и моногенных болезней человека и указывает конкретное моногенное заболевание [3].
1. Прогрессирующие мышечные дистрофии
1.1. Х-сцепленные мышечные дистрофии
1.1.1. Миодистрофия Дюшенна и Беккера (дистрофинопатии — MIM 310 200)
1.1.1.1. Дистрофинопатия у девочек с синдромом Тернера
1.1.2. Миодистрофия Эмери — Дрейфуса (эмеринопатии — MIM 310 300)
1.1.3. Лопаточно-плечевой синдром с деменцией
1.1.4. Миодистрофия Мэбри
1.1.5. Миодистрофия Роттауфа — Мортье — Бейера
1.1.6. Тазово-бедренная миодистрофия Лейдена — Мебиуса
1.2. Аутосомные мышечные дистрофии
1.2.1. Лице-лопаточно-плечевая миодистрофия Ландузи — Дежерина (MIM 158 900)
1.2.1.1. Инфантильная форма лице-лопаточно-плечевой миодистрофии
1.2.2. Скапулоперонеальная миодистрофия Давиденкова (MIM 1 813 500)
1.2.3. Конечностно-поясная миодистрофия (КПМД) Эрба — Рота
1.2.3.1. КПМД 1А (миотилинопатии — MIM 159 100)
1.2.3.2. КПМД 1Е (c парезом гортани, глотки и дистальных мышц)
1.2.3.3. КПМД 2А (калпаинопатии — MIM 253 600)
1.2.3.4. КПМД 2В (дисферлинопатии — MIM 253 601)
1.2.3.5. КПМД 2 с (саркогликанопатии — тяжелая детская аутосомно-рецессивная миопатия — MIM 253 700)
1.2.3.6. КПМД 2 d (саркогликанопатии — тяжелая детская аутосомно-рецессивная миопатия -MIM 600 119)
1.2.3.7. Мышечная дистрофия плечевого и тазового пояса с буллезным эпидермолизом (MIM 226 670)
1.2.4. Миодистрофия Бетлема (MIM 158 810)
1.2.5. Дистальные миодистрофии (MIM 125 660)
1.2.5.1. Дистальная миодистрофия с началом в грудном возрасте
1.2.5.2. Дистальная миодистрофия с началом в детстве
1.2.5.3. Дистальная миодистрофия с поздним дебютом (тип Веландер)
1.2.5.4. Дистальная миодистрофия с ранним началом у взрослых (тип Миоши)
1.2.5.5. Дистальная миодистрофия с накоплением десминовых включений (MIM 601 419)
1.2.6. Окулофарингеальная миодистрофия (MIM 164 300)
1.2.7. Окулярная миодистрофия (прогрессирующая наружная офтальмоплегия Грефе)
2. Врожденные миодистрофии
2.1. Врожденная миодистрофия c поражением головного мозга и глаз (тип Фукуямы — MIM 253 800)
2.2. Врожденная миодистрофия с лейкодистрофией
2.3. Цереброокулярная миодистрофия (MIM 164 300)
3. Спинальные амиотрофии
3.1. Проксимальные спинальные амиотрофии детского возраста
3.1.1. Острая злокачественная инфантильная спинальная амиотрофия Верднига — Гофмана (спинальная амиотрофия 1-го типа) (MIM 253 300)
3.1.2. Хроническая инфантильная спинальная амиотрофия (спинальная амиотрофия 2-го типа) (MIM 253 550)
3.1.3. Ювенильная спинальная амиотрофия (болезнь Кугельберга — Веландер) (MIM 253 400)
3.2. Редкие формы спинальных амиотрофий в детском возрасте
3.2.1. Инфантильная нейрональная дегенерация
3.2.2. Врожденная форма болезни Пелицеуса — Мерцбахера
3.2.3. Врожденная цервикальная спинальная амиотрофия
3.2.4. Атипичный вариант GM-ганглиозидоза
3.2.5. Детский прогрессирующий бульбарный паралич (синдром Фацио — Лонде)
3.2.6. Понтобульбарный паралич с глухотой (синдром Виалетто — Ван Лэре)
3.3. Спинальные амиотрофии взрослых
3.3.1. Бульбоспинальная амиотрофия Кеннеди (MIM 313 200)
3.3.2. Дистальная спинальная амиотрофия
3.3.3. Сегментарная спинальная амиотрофия
3.3.4. Мономиелическая спинальная амиотрофия
3.3.5. Скапулоперонеальная спинальная амиотрофия Старка — Кайзера
3.3.6. Лице-лопаточно-плечевая спинальная амиотрофия Феничела
3.3.7. Окулофарингеальная спинальная амиотрофия
4. Наследственные нейропатии, сопровождающиеся амиотрофиями
4.1. Наследственные мотосенсорные нейропатии
4.1.1. Наследственная мотосенсорная нейропатия, тип 1. Аутосомно-доминантная формa (MIM 118 220, 145 900)
4.1.2. Наследственная мотосенсорная нейропатия, тип 1. Аутосомно-рецессивная форма (MIM 214 400)
4.1.3. Наследственная мотосенсорная нейропатия, тип 2. Аутосомно-рецессивная форма (MIM 601 382)
4.1.4. Наследственная мотосенсорная нейропатия, тип 3. Болезнь Дежерина — Сотта (MIM 3 028 020)
4.1.5. Х-сцепленная наследственная мотосенсорная нейропатия (MIM 302 800)
4.2. Наследственные сенсорные нейропатии
4.2.1. Аутосомно-доминантная сенсорная нейропатия (MIM 162 400)
4.2.2. Наследственная сенсорная и автономная нейропатия, тип 2
4.2.3. Синдром семейной дизавтономии. Синдром Райли — Дея. Наследственная сенсорная и вегетативная нейропатия, тип 3
4.2.4. Врожденная сенсорная нейропатия с ангидрозом. Синдром семейной дизавтономии, тип 2
4.3. Варианты наследственных мотосенсорных нейропатий
4.3.1. Синдром Бернгардта
4.3.2. Синдром Генелла
4.3.3. Синдром Герингама
4.3.4. Синдром Давиденкова
4.3.5. Синдром Штарк — Кезера
4.3.6. Синдром Маватари и Катаямы
4.3.7. Синдром Тевенара (или нейропатия Денни — Брауна)
5. Врожденные структурные миопатии
5.1. Болезнь центрального стержня (MIM 117 000)
5.2. Болезнь множественных центральных стержней
5.3. Врожденная миопатия с диспропорцией типов волокон
5.4. Миотубулярная (центронуклеарная) миопатия (MIM 310 400)
5.4.1. Центронуклеарная миопатия с рецессивным, сцепленным с Х-хромосомой типом
5.4.2. Центронуклеарная миопатия с аутосомно-доминантным типом наследования
5.4.3. Центронуклеарная миопатия с аутосомно-рецессивным типом наследования
5.5. Немалиновая (палочковидная) миопатия
5.5.1. Врожденная быстропрогрессирующая немалиновая миопатия (фатальная)
5.5.2. Врожденная медленнопрогрессирующая немалиновая миопатия (MIM 256 030)
5.5.3. Немалиновая миопатия с дебютом у взрослых
5.6. Врожденная миопатия с внутрицитоплазматическими включениями в виде редуцированных телец
5.7. Миопатия с накоплением телец, сходных с отпечатками пальцев
5.8. Саркотубулярная миопатия
6. Синдром ригидного позвоночника
7. Множественный врожденный артрогриппоз
8. Метаболические миопатии
8.1. Миопатические синдромы при гликогенозах
8.1.1. Дефицит кислой мальтазы (болезнь Помпе)
8.1.2. Дефицит миофосфорилазы (болезнь Мак-Ардла) (MIM 232 600)
8.1.3. Дефицит фосфофруктокиназы (болезнь Тарди)
8.1.4. Дефицит глюкозо-6-фосфатазы (болезнь Гирке)
8.1.5. Дефицит амило-1,6-глюкозидазы (болезнь Кори)
8.1.6. Дефицит альфа-1,4-глюкан-6-глюкозилтрансферазы (болезнь Андерсена)
8.1.7. Дефицит фосфорилазы печени (болезнь Херса)
8.1.8. Дефицит фосфогексоизомеразы (болезнь Сатояши)
8.1.9. Дефицит фосфорилазокиназы печени (болезнь Хьюга)
8.1.10. Дефицит гликоген-синтетазы (болезнь Спенсера — Пита)
8.2. Митохондриальные болезни, обусловленные мутациями митохондриальной ДНК
8.2.1. Синдром Кернса — Сейра (MIM 530 000)
8.2.2. Синдром Пирсона (MIM 557 000)
8.2.3. Синдром МЕLAS (Mitochondrial Encephalomyopathy with Lactic Acidosis and Stroke-Like Episodes) — митохондриальная энцефаломиопатия с лактат-ацидозом и инсультоподобными эпизодами
8.2.4. Синдром МЕRRF (Mioclonic Epilepsy With Ragged Fipes) — миоклонус — эпилепсия с разорванными красными волокнами
8.2.5. Наследственная атрофия зрительных нервов (синдром Лебера)
8.2.6. Синдром NARP
8.3. Митохондриальные болезни, обусловленные мутациями ядерной ДНК
8.3.1. Заболевания, связанные с дефектами дыхательной цепи
8.3.1.1. Дефицит комплекса 1 (NADH: СОQ-редуктаза)
8.3.1.2. Дефицит комплекса 2 (сукцинат-СОQ-редуктаза)
8.3.1.3. Дефицит комплекса 3 (СOQ-цитохром-С-оксидоредуктаза)
8.3.1.4. Дефицит комплекса 4 (цитохром-С-оксидаза)
8.3.1.4.1. Фатальная инфантильная митохондриальная миопатия
8.3.1.4.2. Доброкачественная инфантильная митохондриальная миопатия
8.3.1.4.3. Трихополиодистрофия (болезнь Менкеса)
8.3.1.4.4. Митохондриальные энцефаломиелопатии:
— подострая некротизирующая энцефаломиелопатия (болезнь Лея)
— прогрессирующая склерозирующая полидистрофия (болезнь Альперса)
8.3.1.5. Дефицит комплекса 5
8.3.1.6. Дефицит коэнзима Q
8.3.2. Заболевания, связанные с нарушениями метаболизма молочной и пировиноградной кислот
8.3.3. Заболевания, обусловленные дефектами бета-окисления жирных кислот
8.3.4. Ферментопатии цикла Кребса
8.3.4.1. Дефицит фумаразы
8.3.4.2. Дефицит сукцинатдегидрогеназы
8.3.4.3. Дефицит альфа-кетоглутаратдегидрогеназы
8.3.5. Синдромы дефицита карнитина и ферментов, участвующих в его метаболизме
8.3.5.1. Дефицит карнитин-пальмитилтрансферазы-1 (MIM 255 110)
8.3.5.2. Дефицит карнитин-ацилкарнитин-транслоказы
8.3.5.3. Дефицит карнитин-пальмитоилтрансферазы-2
8.3.5.4. Почечный дефект транспортной системы карнитина
9. Множественный врожденный артрогриппоз
При диагностике прогрессирующих мышечных дистрофий, без сомнения, необходимо исключить воспалительные миопатии, а также разные формы наследственных миотонических и парамиотонических синдромов и миоплегии.
10. Воспалительные миопатии
10.1. Полимиозит
10.2. Дерматомиозит
10.3. Острый инфекционный миозит
10.4. Миозит с включениями телец
10.5. Х-сцепленная вакуольная миопатия
10.6. Гранулематозный миозит
11. Наследственные миотонические синдромы и пароксизмальные миоплегии
11.1. Стационарные, медленнопрогрессирующие миотонии
11.1.1. Врожденная миотония Томсена (аутосомно-доминантный тип наследования) (MIM 160 800)
11.1.2. Врожденная миотония Томсена — Беккера (аутососомно-рецессивный тип наследования) (MIM 255 700)
11.1.3. Приобретенная миотония Тальма
11.1.4. Атрофическая (дистрофическая) миотония, миотоническая дистрофия Россолимо — Куршмана — Штейнерта — Баттена (MIM 160 900)
11.1.5. Миотоническая дистрофия Беккера (аутосомно-рецессивный тип наследования)
11.1.6. Клинические варианты миотонических дистрофий (атрофическая миотония «без атрофий», моносимптомный вариант — миотоническая катаракта, неонатальная форма дистрофической миотонии)
11.2. Периодические (рецидивирующие) формы миотонии
11.2.1. Интемиттирующая миотония Марциуса — Ганземанна (аутосомно-доминантный тип наследования)
11.2.2. Врожденная парамиотония с холодовыми парезами Эйленбурга (аутосомно-доминантный тип наследования)
11.2.3. Врожденная парамиотония без холодовых парезов де Ионга (аутосомно-доминантный тип наследования)
11.2.4. Эпизодическая наследственная миотоническая адинамия Беккера (аутосомно-доминантный тип наследования)
11.3. Наследственные пароксизмальные миоплегии
11.3.1. Гипокалиемическая форма пароксизмальной миоплегии (MIM 170 400)
11.3.2. Гиперкалиемическая форма пароксизмальной миоплегии. Болезнь Гамсторп (MIM 170 500)
11.3.3. Нормокалиемическая форма пароксизмальной миоплегии (MIM 170 600)
И все же детскому неврологу на сегодня прежде всего необходимо знать наиболее часто встречающиеся формы нейромышечных заболеваний.
Миодистрофия Дюшенна является наиболее распространенной формой ПМД. Заболеваемость составляет 13-33 случая на 100 000 родившихся. Тип наследования рецессивный, сцепленный с Х-хромосомой. Гены картированы на коротком плече Х-хромосомы, в 21-м локусе (Хр21). В основе заболевания лежит нарушение синтеза дистрофина. Это субсарколемный мембранный белок с молекулярной массой 427 Кд, состящий из 3685 аминокислот. Данный ген содержит как минимум 5 промоторов, 79 экзонов, состоит из 2,4 млн пар оснований. При миодистрофии Дюшенна уровень дистрофина не превышает 3 % от нормального, тогда как при болезни Беккера он колеблется от 3 до 20 % [3, 6, 7].
Известна также мозговая изоформа дистрофина, экспрессирующаяся в ЦНС [3, 6, 13]. Дебют заболевания — всегда до 5 лет (часто до 3 лет). Характерны жалобы родителей на то, что ребенок часто спотыкается, падает, малоподвижен, предпочитает спокойные игры, ходит на носках. После 6 лет отмечено нарастание мышечной слабости, проксимальной атрофии, ретракции ахилловых сухожилий, кифосколиоза. Способность к самостоятельному передвижению у некоторых сохраняется вплоть до 12 лет, возможность стоять — до 16-летнего возраста. У 60 % больных с миодистрофией Дюшенна имеет место умственная отсталость. Облигатными признаками являются снижение жизненной емкости легких по обструктивному типу, миокардиодистрофии (вносящие существенный вклад в летальность). Больные умирают на 2-3-м десятилетии жизни.
Псевдогипертрофический доброкачественный тип Беккера, относящийся к медленнопрогрессирующим формам ПМД, обусловлен мутацией аллельного гена, локализованного в Хр21 [14, 21]. При данном типе синтез дистрофина полностью не прекращается, синтезируются также другие фракции дистрофина с низкой молекулярной массой. Кроме того, в конце прошлого столетия был открыт и охарактеризован еще один структурный белок мышечной ткани — утрофин, являющийся гомологом дистрофина и кодируемый геном на хромосоме 6q24 [23]. Фенотипические различия между миодистрофиями Дюшенна и Беккера проявляются более поздним дебютом (после 5-10 лет) и мягким течением. Нарушение интеллекта не характерно, ретракции ахилловых сухожилий, кардиальные проблемы не столь выражены. Взрослые больные мужчины через дочь могут передавать заболевание внуку («эффект деда»).
Ранее считали, что МД Дюшенна в 10 % случаев наследуется по аутосомно-рецессивному типу, так как болеют девочки. Однако позднее было установлено, что у девочек с синдромом Тернера (кариотип 45, ХО) МД может развиться, если в большинстве мышечных клеток находится Х-хромосома, несущая мутантный ген МД Дюшенна [21, 23].
На сегодняшний день миодистрофия Дюшенна неизлечима. Наиболее перспективным направлением поиска эффективной терапии считается разработка методов, позволяющих повысить экспрессию дистрофина в скелетных мышцах больных [15]. По общему мнению ученых, именно с генной терапией дистрофинопатий связаны наиболее серьезные надежды добиться уже в обозримом будущем первых реальных результатов в борьбе с этим тяжелейшим заболеванием. Попытки прямого введения в мышцу миобластов (дистрофинпозитивных клеток-предшественников) привели лишь к минимальной и кратковременной экспрессии дистрофина, главным образом вследствие ограниченной миграции вводимых клеток из места инъекции и плохой приживаемости донорских миобластов (Л.П. Горио, С.К. Евтушенко, 1998).
Эмеринопатии — относительно доброкачественные мышечные дистрофии детского возраста. Наследуются по рецессивному, сцепленному с Х-хромосомой типу. В основе болезни лежит нарушение синтеза эмерина, который локализован в мембране ядер мышечных клеток. В эмериновом гене обнаружено 25 мутаций. Мутации вызывают прекращение синтеза эмерина, а иногда — только уменьшение его количества, что приводит к появлению разных фенотипов [7, 17]. Эмеринопатии дебютируют между 4-м и 15-м годами жизни. Чаще всего первым симптомом выступает ходьба на пальцах. Ранними и типичными признаками этой формы являются сгибательные контрактуры в локтевых суставах и разгибателях кисти, ретракции ахилловых сухожилий, затем развиваются слабость и атрофия двуглавых и трехглавых мышц плеча, далее присоединяются мышцы плечевого пояса. Примерно в 20-летнем возрасте наступает относительная стабилизация [5]. Витальный прогноз всецело зависит от степени вовлечения в патологический процесс сердечной мышцы (чаще определяется нарушение сердечной проводимости). Клинически близкой формой является миодистрофия Роттауфа — Мортье — Бейера. Характерной чертой болезни являются быстро прогрессирующие, ранние и выраженные сухожильные ретракции и контрактуры. От МД Эмери — Дрейфуса отличается более диффузным распределением мышечных дистрофий и большей скоростью прогрессирования патологического процесса [5].
Лице-лопаточно-плечевая МД (ЛЛПМД) Ландузи — Дежерина. Болезнь клинически гетерогенна и представлена двумя самостоятельными вариантами. ЛЛПМД-1 — постепенно нисходящий вариант с начальным лице-лопаточно-плечевым фенотипом с последующим вовлечением мышц тазового пояса и бедер. Данный вариант имеет тяжелое течение, и больные могут передвигаться в инвалидных креслах. ЛЛПМД-2 — нисходящий вариант с переходом мышечной слабости от лица, плечевого пояса на перонеальную группу, т.е. с начальным лице-лопаточно-перонеальным фенотипом с последующим вовлечением мышц задней группы бедра, а позднее — тазового пояса (в частности, больших ягодичных мышц) [7, 19, 20].
Дистальные МД характеризуются генетической гетерогенностью. В настоящее время известны аутосомно-доминантные формы с началом в грудном, детском и зрелом возрасте. Аутосомно-рецессивная форма (дистальная миодистрофия Миоши) начинается в подростковом возрасте или позже и связана с хромосомным локусом 2р12-14. Дистальная миодистрофия с началом в детском возрасте дебютирует в первые 2 года жизни. Генетическая природа неясна. Ведущими симптомами являются шлепающие стопы, слабость мышц — разгибателей кисти. Течение доброкачественное, медленно прогрессирующее. Дистальная миодистрофия с началом в детстве дебютирует после 4 лет. Ген картирован на 14-й хромосоме. Симптоматика поражения мышц дистальных отделов конечностей. Течение благоприятное. Дистальная миодистрофия типа Миоши дебютирует между 15-м и 25-м годами жизни. Вначале слабость и атрофия имеют место в икроножных мышцах, передняя группа мышц голеней остается интактной. Ахилловы рефлексы отсутствуют, все остальные вызываются. Медленно прогрессирующее течение [6].
Скапулоперонеальная МД Давиденкова — аутосомно-доминантная форма мышечной дистрофии. Предполагается, что заболевание обусловлено мутацией гена на 12-й хромосоме. Слабость перонеальной и плечевой мускулатуры проявляется в различной последовательности или одновременно. Имеет мягкое течение. Кардиомиопатии нетипичны.
Врожденные МД (ВМД) клинически напоминают врожденные миопатии с характерными морфологическими изменениями мышц. Они возникают внутриутробно или в первые месяцы жизни и проявляются задержкой моторного развития, диффузной мышечной гипотонией, ранними контрактурами мышц и деформацией суставов. Однако в отличие от врожденных миопатий ВМД быстро прогрессируют (имеются в виду аутосомно-рецессивные формы), так что в возрасте 4-8 мес. дети становятся обездвиженными из-за слабости и контрактур. Помимо этого, при ВМД в отличие от врожденных миопатий отсутствуют характерные морфологические изменения в скелетных мышцах, выявляемые при световой и электронной микроскопии. Описаны классические аутосомно-рецессивные тяжелые формы ВМД с дефицитом мерозина и доброкачественные ВМД, мерозинпозитивные, с дефицитом интегрина или актинина. ВМД представляют клинически и генетически гетерогенную группу мышечных болезней [18].
ВМД Бетлема — доброкачественная болезнь, характеризующаяся началом в раннем детстве, доброкачественным течением с нормальной продолжительностью жизни, конечностно-поясным типом распределения амиотрофий, сохранностью лицевых мышц, контрактурами в межфаланговых, локтевых и голеностопных суставах.
Другая группа ВМД обусловлена сочетанным поражением мышц, головного мозга и сетчатки глаза. Можно сказать, что это энцефаломиопатии. Наиболее изучен тип Фукуяма — врожденная миодистрофия, сопровождающаяся церебральной дисплазией и гипоплазией. Болезнь генетически и клинически гетерогенна. Среди детей неяпонского происхождения патология мозга, глаз и мышц встречается при цереброокулярной миодистрофии. Гипотония и генерализованная слабость выявляются с рождения. Тяжелое психическое отставание определяется на 1-м году жизни. По данным МРТ могут обнаруживаться аномалии формирования извилин, гетеротопия нейронов, дисмиелинизация. В большинстве случаев представлена гидроцефалия (синдром Варбурга). Патология глаз включает помутнение роговицы, катаракту, дисплазию сетчатки или ее отслойку, гипоплазию зрительных нервов [7, 16].
Наследственные полинейропатии (невральные амиотрофии) — обширная группа гетерогенных заболеваний, поражающих периферическую нервную систему. Болезнь Шарко — Мари — Тута — наиболее распространенная форма наследственных нейропатий, относящаяся к группе наследственных моторно-сенсорных нейропатий (НМСН). Существует несколько вариантов этого заболевания.
1-й тип (НМСН-1) — группа заболеваний, вызванных генетическим дефектом синтеза периферического миелина (демиелинизирующий, гипертрофический вариант болезни Шарко — Мари — Тута). Наиболее часто НМСН-1 наследуются по аутосомно-доминантному типу. Генетически выделяют не менее трех вариантов НМСН-1 с этим типом наследования. Наиболее распространенный вариант — НМСН-1А, при котором генетический дефект выявлен на коротком плече 17-й хромосомы и представляет собой удвоение или точечную мутацию гена, кодирующего синтез белка периферического миелина. При другом, реже встречающемся варианте (НМСН-1В), который клинически и электрофизиологически неотличим от НМСН-1А, генетический дефект обнаружен на длинном плече 1-й хромосомы, связан с точечной мутацией гена, кодирующего белок миелин. В ряде случаев генетический дефект остается неизвестным (НМСН-С). Реже НМСН-1 наследуются по аутосомно-рецессивному типу, в этих случаях выявлена мутация на длинном плече 8-й хромосомы [12].
Симптомы заболевания обычно проявляются на 1-м десятилетии жизни. Дети предъявляют жалобы на боли в мышцах голени, возникающие после физической нагрузки, затруднения при беге или подъеме по лестнице, утомляемость, частые падения. Чаще дети ходят с упором на передние отделы стоп. Патологический процесс имеет восходящий тип поражения. Раньше других страдает вибрационная и тактильная чувствительность. Характерна полая стопа с молоточкообразной деформацией пальцев стопы с формированием стопы Фридрейха. Течение заболевания медленно прогрессирующее [12].
НМСН 2-го типа имеют название нейрональной, или аксональной, формы болезни Шарко — Мари — Тута, патоморфологически проявляющейся признаками аксональной дегенерации и вторичной демиелинизации без образования «луковичных головок». НМСН-2 передаются по аутосомно-доминантному типу, реже — по аутосомно-рецессивному. При обследовании семей с НМСН-2А аутосомно-доминантным типом наследования ген выявляется на коротком плече 1-й хромосомы, в других случаях — на длинном плече 3-й хромосомы (НМСН-2В). Выделяют форму НМСН-2С, не связанную с локусом 1-й или 2-й хромосомы. НМСН-2D вызываются мутацией в коротком плече 7-й хромосомы. НМСН-2 наследуются по аутосомно-рецессивному типу, обычно протекают более тяжело и проявляются в первые годы жизни, гены картированы на длинном плече 8, 11, 5-й хромосом. Клинически заболевание напоминает НМСН 1-го типа, но проявляется позднее (13-16 лет), реже вовлекаются руки, менее выражены признаки нарушения чувствительности и деформации стопы. Одним из главных критериев, помогающим отдифференцировать НМСН 2-го типа от НМСН 1-го типа, является ЭНМГ: при НМСН-2 нет существенного снижения скоростей распространения возбуждения, отмечается выраженное или даже полное отсутствие М-ответа [12].
В числе самых распространенных наследственных заболеваний аутосомно-рецессивной природы находится спинальная мышечная атрофия (СМА). Частота встречаемости колеблется в среднем 1 : 10 000 новорожденных. СМА характеризуется дегенерацией тел двигательных нейронов передних рогов спинного мозга, которая приводит к прогрессирующему параличу конечностей и туловища, ассоциированному с мышечной атрофией. Выделяют 3 типа СМА с увеличением возраста начала и уменьшением тяжести заболевания от 1-го к 3-му типу. СМА 1-го типа (тяжелый тип, болезнь Верднига — Гоффмана) характеризуется возрастом начала заболевания от рождения до 6 мес. Больные неспособны сидеть самостоятельно, умирают обычно в первые 2 года жизни. СМА 2-го типа (промежуточный тип) характеризуется возрастом начала заболевания до 18 мес., больные могут сидеть, но не могут стоять или ходить без поддержки. Для СМА 3-го типа (мягкий тип, болезнь Кугельберга — Веландера) характерным является возраст начала заболевания после 18 месяцев; больные длительное время сохраняют способность стоять и ходить. Локус аутосомно-рецессивной СМА картирован в хромосомном регионе 5q11.2-13 [12].
Таким образом, с учетом наличия клинической и генетической гетерогенности МД, возможности применения в будущем генной терапии можно считать, что МД будут классифицироваться по генному продукту, лежащему в основе заболевания. Это должно стать фундаментом для следующего важнейшего шага — разработки эффективных методов лечения ПМД. Точная нозологическая диагностика МД невозможна без иммунобиохимических, иммуногистохимических и молекулярно-генетических исследований [6]. Однако, учитывая ограниченный доступ к проведению массового скрининга (сегодня исследования проводятся в Институте молекулярной биологии и генетики Украины, г. Киев, клинике «Исидa IVF», г. Киев, государственном учреждении МГЦ, г. Москва), социально-экономическое положение большей части пациентов, включение в разработку паттерна достоверной диагностики в настоящее время всеже не представляется возможным. Поэтому на данном этапе для диагностики ранних проявлений прогрессирующих мышечных дистрофий и амиотрофий, их фенокопий сотрудниками кафедры детской и общей неврологии ФИПО ДонНМУ и сотрудниками ДОДКЦНР разработан диагностический и лечебный паттерн, адаптированный к практическому здравоохранению, который включает в себя:
-
Клинический осмотр с использованием компьютерной диагностической программы «Нейромиоген» (авторы: С.К. Евтушенко, И.А. Садеков, 1995).
-
Биохимическое исследование ферментов крови (АЛТ, АСТ, КФК, ЛДГ) на аппарате «Фотометр-КФК-3»; исследование иммунологического статуса (Ts, Th, T, CD25, CD4/CD8).
-
Электронейромиографию с помощью компьютерного электромиографа «Феникс-241» фирмы «НейроТех» (Россия) — используются стандартные накожные регистрирующие электроды, стимулирующие биполярные электроды; при электромиографии — концентрические игольчатые электроды, включая одноразовые.
-
Электрокардиографию (аппарат «ЭК1К-01»), при необходимости — холтеровский мониторинг ЭКГ с измерением АД.
-
ЭхоКГ (аппарат «Logic 200 ProSeries»).
-
Исследование функции внешнего дыхания с помощью аппарата «Спиросефт-3000» (Япония).
-
Консультации ортопеда, педиатра, кардиолога, иммунолога, окулиста, психиатра, психолога.
-
При необходимости — дифференциальную диагностику системных заболеваний, проявляющихся амиотрофическим синдромом, включая биопсию мышц.
-
Обследование в областном медико-генетическом центре.
-
При затруднении постановки диагноза показано МРТ головного, спинного мозга (аппарат «Gyrocsan Intera T10» Голландия).
-
Для исключения сосудистых аномалий показано проведение УЗДГ (аппарат «Logidop-4» фирмы «Kranzbuhler»), цветное дуплексное сканирование (аппарат «Sanoline Elegre advanced», Siеmens).
-
Денсинометрию костей.
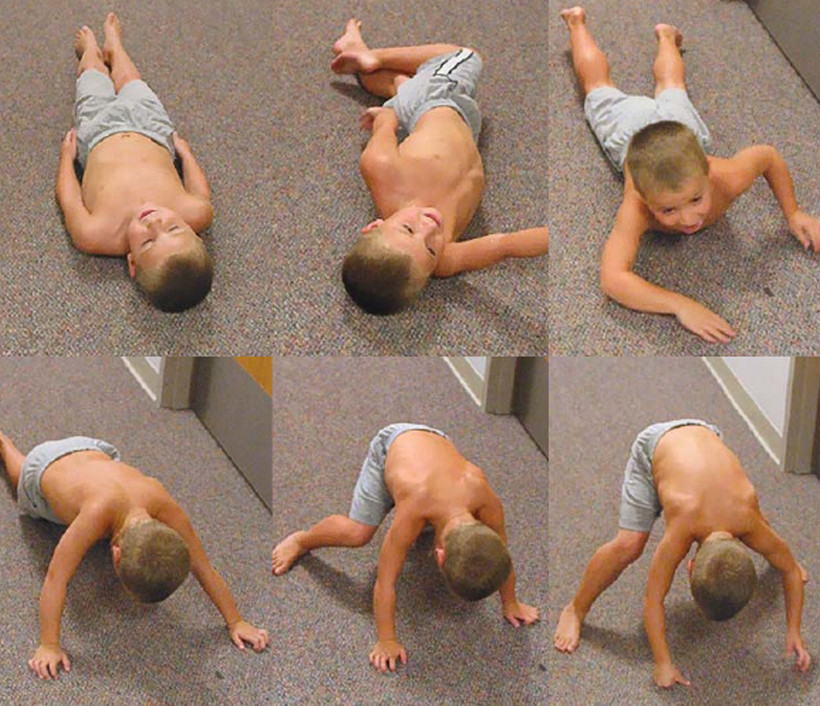
Мышечная слабость и утомляемость — эти два симптома следует идентифицировать. Жалобы на утомляемость без признаков мышечной слабости чаще свидетельствуют о тревожности, церебрастеническом синдроме, эндокринном или системном заболевании. Для того чтобы отличить эти два симптома, необходимо оценить походку, попросить пройтись на носках, пятках, поприседать, встать с пола из положения лежа на животе, спине, сжать пальцы врача, поработать руками, поднятыми выше уровня плеч. Трудности при выполнении данных силовых упражнений, вероятнее всего, больше свидетельствуют в пользу мышечной слабости, чем в пользу утомляемости. Для уточнения распространенности мышечной слабости (ограниченной или генерализованной) и степени ее выраженности последовательно исследуют основные группы мышц. Оценка степени мышечной слабости с использованием стандартной шкалы позволяет точнее определить выраженность, локализацию мышечной слабости, сравнить ее при повторных исследованиях.
Наиболее широко в настоящее время миологами используется шкала определения степени мышечной слабости, разработанная американскими учеными (R.D. Adams, F.L. Mastaglia, N.G. Lain, 2005): 0 — паралич; 1 — минимальные сокращения; 2 — активное движение при устранении силы тяжести; 3 — слабое сокращение против силы тяжести; 4 — активное движение против силы тяжести и сопротивления; 5 — нормальная сила.
Промежуточные состояния оценивают знаками «+» или «-«. Например, мышечную слабость чуть меньшую, чем 4 балла, обозначают «4+», а чуть большую — «4-«. Использование промежуточных значений увеличивает число пунктов в шкале до 10.
Симптомокомплекс «вялого ребенка» наблюдается при широком спектре заболеваний нервно-мышечной системы, перинатальных поражениях ЦНС, заболеваниях соединительной ткани (V. Dubowitz, 1986) [2]. Поэтому очень важно знать дополнительные симптомы, позволяющие заподозрить ту или иную форму нейромышечной патологии.
Изложенные выше данные имеют большое диагностическое значение. Учитывая дебют заболевания, возраст пациента на момент развития заболевания, динамику, преимущественную вовлеченность в патологический процесс мышц, локализацию мышечной слабости, наличие или отсутствие наследственной отягощенности, знание основных синдромов дает возможность определить или заподозрить все наиболее распространенные мышечные заболевания.
ЭМГ помогает установить диагноз, в особенности отличить денервационную атрофию от миопатии. Электрофизиологические тесты позволяют легко дифференцировать причину мышечной слабости. Существуют две стандартные методики исследования: 1) ЭМГ, при которой игольчатые электроды вводятся в мышцы, что позволяет регистрировать спонтанную активность (потенциалы фибрилляции, потенциалы фасцикуляции, положительные острые волны, миотонические разряды, псевдомиотонические разряды), изменение параметров потенциалов двигательных единиц (ПДЕ).
Для миопатического типа поражения характерно:
-
уменьшение средней длительности ПДЕ (на 21 % и более);
-
снижение амплитуды ПДЕ (100-300 мкВ при норме 350-600 мкВ);
-
повышение фазности потенциалов (60-70 % при норме до 5 %), выраженная полифазия (до 90 %) отмечается при злокачественных формах ПМД (формы Дюшенна, Эрба — Рота);
-
спонтанная активность регистрируется преимущественно в виде потенциалов фибрилляции (ПФ);
-
наибольшая выраженность спонтанной активности в виде потенциалов фибрилляции отмечается при быстропрогрессирующих формах миодистрофии (формы Дюшенна, Эрба — Рота и др.).
Для нейронального (переднерогового) типа поражения характерно:
-
увеличение длительности и амплитуды ПДЕ, причем у пациентов со спинальной амиотрофией Верднига — Гоффмана было выявлено значительное увеличение длительности (40 % и более), амплитуды ПДЕ (до 3000-5000 мкВ при норме 350-600 мкВ);
-
доли полифазных потенциалов (до 90 %);
-
спонтанная активность регистрируется преимущественно в виде отдельных потенциалов фасцикуляции (ПФЦ).
У больных со спинальной амиотрофией Верднига — Гоффмана выявляется бурная спонтанная активность преимущественно в виде потенциалов фасцикуляции.
Для неврального типа поражения характерно:
-
увеличенние амплитуды ПДЕ (до 1500-3000 мкВ);
-
увеличение длительности (на 15-40 %);
-
увеличение числа полифазных ПДЕ (60-80 %);
-
спонтанная активность в виде положительных острых волн;
-
чрескожная стимуляция периферических нервов с регистрацией моторных и сенсорных потенциалов действия (исследование скорости проведения возбуждения по двигательным и чувствительным волокнам) и определение амплитуды СМПД, дистальной латентности.
И все же наиболее информативной методикой является биопсия мышцы и нервов, которая в ряде случаев позволяет выявить специфические особенности заболевания. Вместе с тем нельзя закрывать глаза на проблему, обусловленную отсутствием клинико-патоморфологических миологов, которые компетентно могут дать заключение о характере поражения мышц. Миопатии и миодистрофии характеризуются диффузной потерей мышечных волокон, которые замещаются жировой или соединительной тканью. При полимиозите появляются признаки воспалительных изменений. Денервационные атрофии характеризуются выраженным уменьшением размеров мышечных волокон в пораженных двигательных единицах и увеличением интактных двигательных единиц. Избыточное накопление липидов или гликогена в сохранных мышечных волокнах позволяет заподозрить липидные и гликогеновые болезни накопления. Митохондриальные заболевания различных типов можно обнаружить при окраске препаратов трихромом по Гомори, позволяющей определить скопления митохондрий, которые окрашиваются в красный цвет. С помощью световой и электронной микроскопии можно выявить демиелинизацию, формирование луковицеобразных утолщений нервного ствола, аксональную дегенерацию, валлеровское перерождение [24].
К сожалению, с каждым годом идентификация ранних форм нейромышечных заболеваний увеличивается, что и подтверждается нашими наблюдениями.
Таким образом, в 1998 г. было идентифицировано 5 форм нейромышечной патологии; в 1999 г. — 10 форм; в 2000 г. — 12 форм; в 2006 г. — 17 форм.
Число детей с нейромышечной патологией, получающих курс реабилитационного лечения в Центре, ежегодно возрастает. В 2006 году 158 детей (от 6 мес. до 16 лет) проходили курс лечения (у 25 детей впервые диагностирована нейромышечная патология, 133 ребенка регулярно получают лечение и находятся под наблюдением сотрудников Центра). При этом 129 детей — из Донецкой области, 29 — из других областей Украины и стран СНГ.
10-летний опыт работы с больными с нейромышечной патологией свидетельствует о том факте, что ранняя, своевременная диагностика — это надежда на продление жизни больного. Определение формы нейромышечной патологии (мышечная дистрофия, спинальная либо невральная амиотрофия), выявление сопутствующей патологии (кардио-, пневмопатии, ортопедические проблемы, когнитивные нарушения) позволяет подобрать патогенетически обоснованную терапию, косвенным образом воздействуя на определенные звенья патогенеза и улучшая, таким образом, качество жизни больного.
На основе мировых, отечественных и собственных данных в Центре разработаны стандарты лечения. Лечение, которое проводится больным, является комплексным и включает патогенетическую, специальную медикаментозную терапию с учетом степени тяжести заболевания (легкая, средняя, тяжелая), стадии (компенсации, субкомпенсации, декомпенсации), физиотерапевтические процедуры, синглетно-кислородную терапию, лечебную физкультуру (дыхательная гимнастика, стренч-гимнастика), специальную диету, электроакупунктуру, щадящий массаж функционально сохранных мышц.
-
Сбалансированное лечебное питание (продукты, содержащие белок, полиненасыщенные жирные кислоты, витамины, микроэлементы): овощи, творог, рыба, печень, соевое мясо. Весной и осенью — курсовой прием поливитаминных препаратов и микроэлементов (активал, мультитабс, биовиталь, мильгамма, нейровитан, неуробекс).
-
Медикаментозное лечение прогрессирующих мышечных дистрофий.
Медикаментозное лечение назначается с учетом полученных результатов клинико-инструментального исследования и сопутствующей патологии (кардио-, пневмопатии). При доброкачественных формах ПМД (Говерса — Веландера, Давиденкова, Бетлема, Беккера, Эмери — Дрейфуса) в стадии стойкой компенсации, при легкой, легко-средней степени тяжести заболевания целесообразно назначение курсами препаратов «метаболического» действия, направленных на улучшение, поддержание обменных, «энергетических» процессов неповрежденных миоцитов, кардиомиоцитов. К таким препаратам относятся: АТФ-лонг, цитофлавин, кардонат, элькар, милдронат, магнерот, витамин Е, метионин. Назначается по 1-2 препарата курсами 2-3 раза в год.
При наличии жалоб на боли в мышцах нижних конечностей, чувство «стягивания» мышц особенно хорошо зарекомендовал себя цитрулина малат. Препарат способствует «утилизации» молочной кислоты, одновременно обладая метаболическими свойствами. Данный препарат широко используют в профессиональном спорте при физическом перенапряжении, перед соревнованиями.
При доброкачественных формах в стадии субкомпенсации, при средней степени тяжести заболевания, на ранних стадиях патологического процесса в стадии компенсации при злокачественных (быстро прогрессирующих) формах ПМД (Дюшенна, Эрба — Рота) назначается 10% раствор карнитина хлорида (данный препарат обладает метаболическим, нейротрофическим, кардиотрофическим и антиоксидантным свойствами) c кокарбоксилазой, аскорбиновой кислотой в/в капельно на фоне в/м введения пиридоксина гидрохлорида и перорального приема метионина. 3-4 курса № 10 в год с дальнейшим переходом на пероральный прием препарата кардонат в амбулаторных условиях длительностью до 2 мес.
У детей до 5 лет предпочтение отдается жидким формам карнитинсодержащих препаратов ввиду удобства применения и лучшей переносимости.
При этих же формах для фармакопунктур используются антигомотоксические препараты, препараты нейротрофического действия (церебрум композитум, траумель и др.).
При наличии церебрастенического, амиотрофического синдрома назначается мильгамма по 1-2 мл в/м через день № 10 с дальнейшим переходом на прием препарата внутрь в виде драже до 1 мес.
На стадии развернутой клинической картины миодистрофии Дюшенна назначается иммуноглобулин человека нормальный в дозе 5,0-7,0 мл/кг на инфузию . Весь объем иммуноглобулина растворяется в 4 раза изотоническим раствором и вводится со скоростью 15 капель в минуту. Количество инфузий — от 3 до 5. На фоне малых доз преднизолона по 5 мг 1 раз в день 3-6 мес. Иммуноглобулин продемонстрировал достаточно высокую эффективность, было отмечено снижение показателей КФК, ЛДГ, АЛТ, АСТ в среднем на 20 %, дети отмечали нарастание силы, переносимости физических нагрузок. Эффективность иммуноглобулинов при миодистрофии Дюшенна нами объясняется следующим образом:
-
Не исключено, что разрушение дистрофинассоциированного комплекса белков может происходить не только в результате генетических факторов, но и вследствие вирусных инфекций. Это, в частности, доказано в отношении вируса Коксаки, способного разрушать дистрофин и ассоциированный с ним комплекс белков в кардиомиоцитах.
-
Распад мышечных волокон способствует развитию апоптоза, что подтверждают наши исследования по высокому уровню СD25 (маркер апоптоза). Некроз миоцитов, распад белков дистрофинассоциированного комплекса (белки цитоскелета, трансмембранные белки, периферические мембранные белки) с выбросом их фрагментов в кровь, приводящие к продукции циркулирующих иммунных комплексов и развитию аутоиммунизации. При злокачественных формах мышечных дистрофий в стадии субкомпенсации, начальных проявлениях декомпенсации, в стадии выраженных клинических проявлений при доброкачественных формах используем неотон (фосфокреатинин, «супер-АТФ») в/в капельно 1,0-4,0, № 5, 3-4 курса в год. Данный препарат используется у взрослых в комплексном лечении инфаркта миокарда, мы же стали его использовать для увеличения сократительной способности миокарда.
Кроме того, этим же детям назначаются подддерживающие дозы преднизолона 5-10 мг утром 1-3 месяца (курсами).
-
Медикаментозное лечение спинальных и невральных амиотрофий.
В лечении спинальных амиотрофий целесообразно назначение миелотоников, препаратов нейротрофического действия, улучшающих метаболизм и микроциркуляцию спинного мозга.
Мы используем церебролизин, актовегин (преимущественно в виде фармакопунктур); цераксон по 2 мл 2-3 раза в день внутрь до 45 дней; нуклео ЦМФ форте в/м с дальнейшим переходом на прием внутрь, мильгамму в/м. Амбулаторно назначаются традиционные препараты метаболического действия: триметабол, кардонат, АТФ-лонг, фолиевая кислота, витамины Е и А, метионин, нейровитан, никотиновая кислота, тиоцетам, пирацетам.
В определении тактики лечения невральных амиотрофий помогают ЭМГ-данные, позволяющие установить преимущественный тип поражения нервного волокна (аксональный, демиелинизирующий, смешанный). При обнаружении миелинопатии целесообразно назначение мильгаммы, входящий в состав данного препарата бенфотиамин увеличивает биодоступность в ткани, пиридоксин усиливает регенеративные процессы в нервной ткани (дозировки: детям с 5 лет до 8 лет — по 0,5-1 мл в/м № 10 через день, с 8 до 15 лет — по 1-2 мл в/м № 10 через день с дальнейшим переходом на пероральный прием по 1 драже 2-3 раза в день); нуклео ЦМФ форте, проявляющий трофические свойства за счет обеспечения фосфатных групп, необходимых для формирования сфингомиелина и глицерофосфата — основных компонентов миелиновой оболочки (дозировки: детям от 1 года до 3 лет — по 1/4 мл в/м 1 раз в день № 6, от 3 до 7 лет — 1/2-1 мл в/м 1 раз в день № 6, старше 7 лет — по 2 мл 1 раз в день № 6 с дальнейшим переходом на пероральный прием в виде капсул по возрастным дозировкам), в промежутках между приемами назначаем семакс 0,1%, брэйн комплекс, тиоцетам.
При выявлении аксонального типа поражения используем нейромидин 0,5%, 1,5% в/м № 20 с дальнейшим переходом на пероральный прием, цераксон (раствор для приема внутрь) и нейромедин 1 табл. 2 раза в сутки
-
Коррекция ортопедических проявлений составляет весомую часть в реабилитации больных с нейромышечной патологией.
Наиболее часто в клинике преобладают:
-
деформация туловища, позвоночника (нейромышечный сколиоз), конечностей;
-
врожденный вывих и подвывих тазобедренных суставов, контрактуры суставов, мышц;
-
деформация стоп (эквиноварусные стопы, косолапость, плоскостопие, деформация стоп по типу Фридрейховских, гипермобильность суставов, нестабильность надколенника, сухожильные ретракции, псевдогипертрофии мышц) [8].
По нашим наблюдениям, до 40 % детей, у которых первично выявлена нейромышечная патология и в клинике которых имеет место симптоматика поражения опорно-двигательного аппарата, длительно наблюдались и лечились у ортопедов по месту жительства. Поступают такие дети в стадии выраженных клинических симптомов болезни, когда существенно повлиять на течение патологического процесса практически невозможно.
Лечение ортопедических проявлений у больных с МД является сложной и до конца не решенной проблемой, анализ литературы по этому поводу свидетельствует о существовании разных точек зрения. Мы используем следующую методику: при начальных проявлениях контрактур, ретракции сухожилий используются щадящий массаж с применением трофических мазей, фармакопунктуры с цель Т, траумель С, направленные на улучшение питания сустава, специально разработанные методики ЛФК, фиксацию конечности в положении достигнутой коррекции контрактуры суставов, шины, валики для профилактики контрактур, фиксацию конечностей в физиологическом положении на ночь с использованием туторов; с целью адаптации передвижения с оптимальной коррекцией деформаций используются стельки, ортопедическая обувь, надколенники. При доброкачественных формах МД в стадии компенсации возможно проведение оперативного вмешательства, направленного на предупреждение и избавление контрактур, сухожильных ретракций, коррекцию деформаций
В целях укрепления мышечного корсета спины используется фармакопунктура паравертебрально с применением нейромидина, церебролизина, актовегина, кортексина, цианокобаламина. В стадии субкомпенсации мы рекомендуем 1-2-часовое ношение реклинаторов, корсетов в моменты наибольшей нагрузки на позвоночный столб (сидение, ходьба и др.), несмотря на категорическое несогласие с этим ортопедов. В стадии декомпенсации, тяжелой степени рекомендуется практически постоянное ношение корсетов, так как в данной стадии заболевания происходят выраженная атония и гипотрофия мышечного корсета, влекущие за собой резкую деформацию позвоночного столба, приводящую к вторичной висцеропатии, ухудшению работы сердца, легких, влекущих за собой еще большую декомпенсацию патологического процесса.
Наличие остеопороза у большинства больных с миопатиями и амиотрофиями обусловливает назначение кальцийсодержащих препаратов: кальций D3 (кальцемин адванс) курс 30-60 дней, 3-4 курса в год под обязательным контролем Са крови, денсинометрией костей, УЗИ почек.
-
Одним из ведущих механизмов, приводящих к глубокой инвалидизации больных с НМЗ, является слабость дыхательной мускулатуры, приводящая к нарушению грудной экскурсии, дыхание становится поверхностным.
В 2006 году вентиляционную функцию легких и биомеханику дыхания исследовала у 30 больных врач кабинета функциональной диагностики Л.Ф. Евтушенко (в стадии развернутых клинических проявлений), из которых 18 больных — с ПМД Дюшенна, 1 больной — с ПМД Эрба — Рота, 5 детей — с ПМД Эмери — Дрейфуса, 2 ребенка — с ПМД Говерса — Веландера, 3 больных — со спинальной амиотрофией Кугельберга — Веландера, 1 больной — с дистальной спинальной амиотрофией. Исследование проводилось на аппарате «Спиросефт-3000» (Япония). У всех детей был выявлен обструктивный тип нарушения вентиляционной функции легких, обусловленный внелегочными причинами. Сопутствующая патология со стороны опорно-двигательного аппарата — деформация туловища, позвоночника (нейромышечный сколиоз) — увеличивает тяжесть дыхательных расстройств. По данным пневмотахиметрии у этих детей выявляется обструктивный тип нарушения вентиляционной функции легких, уменьшение жизненной емкости легких. Сопутствующая кардиальная патология (миокардиодистрофии, функциональная кардиопатия, диспластическая кардиопатия) приводит к нарушениям в малом круге кровообращения, что проявляется ухудшением микроциркуляции легких, что, в свою очередь, приводит к явной или скрытой хронической дыхательной недостаточности. Перечисленные причины, а также сниженная иммунная реактивность организма, церебрастенический, панастенический синдромы негативно сказываются на течении заболевания, порой приводят к «сбою» компенсаторных механизмов и как исход — к декомпенсации патологического процесса.
Поэтому необходимо назначение препаратов, влияющих непосредственно на иммунокомпетентные клетки и центральные механизмы регуляции иммунитета, через которые оказывается вторичное иммуностимулирующее и энерготонизирующее влияние на организм. К таким препаратам относятся: бронхомунал-П, рибомунил, иммунал, иммунотон, ИРС.
Из немедикаментозных методов такими свойствами обладает Valkion-терапия. Физико-химическая концепция Valkion-терапии базируется на фотохимической сенсибилизации воздуха и воды с образованием вторичных долгоживущих физиологически активных форм кислорода и оксида азота — Valkion-факторов. Кроме того, патогенетический эффект Valkion-терапии включает в себя активацию клеточного метаболизма, снижение гипоксии тканей, нормализацию реологических свойств крови, восстановление слизистой бронхов, нормализацию функции внешнего дыхания, улучшение дренажной функции бронхов, положительные изменения динамики ЭКГ, снижение уровня молочной кислоты в мышцах.
Мы используем следующую схему: 1-й день — 100 мл воды, 5 мин ингаляции, 2-3-й день — 150 мл воды, 9 мин ингаляции, 4-й и последующие дни — 200 мл воды, 14 мин игаляции. 3-4 курса № 15 в год.
Детей и их родителей обучают специально разработанной в нашем Центре дыхательной гимнастике, в основе которой лежит методика успокаивающего нижнего дыхания, укрепляющего среднего дыхания, гармоничного полного дыхания, ритмичного дыхания, энергетизирующего дыхания, стимулирующего дыхания, очищающего «ха-дыхания», вокалотерапии (произношение звуков во время активного выдоха) с особым акцентом на гласные звуки. С целью повышения эффективности звуковая гимнастика сочетается с вокалотерапией. В домашних условиях рекомендуются использование музыкальных игрушек, детских музыкальных инструментов, занятия музыкой, вокалом, надувание резиновых шаров.
-
Когнитивные нарушения, в частности, при ПМД Дюшенна имеют место в 40 % случаев. Наряду с традиционными ноотропными препаратами (церебролизин, тиоцетам, пирацетам, луцетам и др.) мы предпочитаем назначение и таких ноотропных препаратов, как семакс 0,1% по 3-4 капли в носовые ходы 2 раза в день 10 дней, чередуя с приемом когитума по 1/2-1 ампулы внутрь 2 раза в день 10 дней, курсами цераксон по 2 мл 2-3 раза в день — 45 дней.
-
Проблема поражения сердечно-сосудистой системы при нейромышечных заболеваниях актуальна, поскольку летальный исход большинства из них обусловлен именно вовлечением сердца в патологический процесс, так как сердечная мышца поражается первично вследствие дефицита того же структурного компонента, которого нет и в скелетной мышце. Сердце — непрерывно и интенсивно работающий орган, и нередко дебют миопатии начинается именно с кардиологических проявлений, в то время как симптомы миопатического процесса могут быть маскированными [9].
Согласно мировым стандартам эхокардиографического исследования, выделяют 6 клинико-патогенетических стадий поражения сердечно-сосудистой системы при нейромышечных заболеваниях [9]. На основе стандартов и собственных наблюдений за 2006 год было обследовано 150 детей с нейромышечными заболеваниями. ЭхоКГ проводилось на аппарате «Logic 200 Proseries» (врач Д.А. Сохань).
-
Диастолическая дисфункция миокарда по рестриктивному типу на стадии начальных проявлений заболевания (уменьшение скорости потока крови на митральном клапане с уменьшением амплитуды предсердной волны, уменьшение диастолического диаметра и объема левого желудочка, умеренное снижение фракции выброса левого желудочка) — 18 детей.
-
Миокардиальная стабилизация на стадии компенсации заболевания (нормализация допплерэхокардиографических показателей) — 42 ребенка. На данных стадиях целесообразно применение кардиотрофической терапии перорально 2-3 курса в год (АТФ-лонг, рибоксин, милдронат, кардонат, элькар, калия оротат, магнерот).
-
Систолическая дисфункция миокарда на стадии субкомпенсации миопатического процесса (умеренное увеличение систолического диаметра и объема левого желудочка, умеренное уменьшение фракции выброса) — 46 детей.
-
Миокардиальная псевдостабилизация на стадии декомпенсации-А миопатического процесса (нормализация допплерэхокардиографических показателей) — 30 детей.
-
Гипокинезия левого желудочка на стадии декомпенсации-В (выраженное увеличение систолического диаметра и объема левого желудочка, значительное снижение фракции выброса) — 12 детей.
-
Дилатация левого желудочка на стадии декомпенсации-С (значительное увеличение диастолического и систолического диаметра и объема левого желудочка, резкое снижение фракции выброса) — 2 ребенка.
Применение расчетных эхокардиографических параметров позволяет более тонко разобраться в патогенезе заболевания, констатировать предпатологические изменения и заблаговременно начать лечение. Такая тактика позволяет продлить срок самостоятельной ходьбы больных, отсрочить время наступления манифестной сердечной недостаточности (врач-педиатр, кардиолог Е.В. Дегонская).
На 3-4-й стадиях целесообразно применение 10% раствора карнитина хлорида (данный препарат обладает метаболическим, нейротрофическим, кардиотрофическим и антиоксидантным свойствами) c кокарбоксилазой, аскорбиновой кислотой в/в капельно на фоне в/м введения пиридоксина гидрохлорида и перорального приема метионина. № 10, 3-4 курса в год. Амбулаторно дети принимают кардонат по 1 капсуле 2-3 раза в день — 45 дней.
На 4-6-й стадии используется неотон (фосфокреатинин, «супер-АТФ») в/в капельно № 5, 3-4 курса в год.
При анализе результатов реабилитационного лечения детей с нейромышечной патологией отмечена положительная динамика, которая выражается в том, что у больных со злокачественными формами НМЗ (ПМД, форма Дюшенна, Эрба — Рота) по сравнению с детьми с аналогичными формами, когда в Центре не было специализированных коек, на 2-3 года дольше сохраняется способность к самостоятельному передвижению, позднее формируются контрактуры, реже протекает ОРВИ, длительное время поддерживается насосная и сократительная способность миокарда. Дети с доброкачественными формами НМЗ долгое время сохраняют вертикальную позу, социально адаптированы. Увеличилась и продолжительность жизни, поскольку клинические проявления сопутствующей патологии в связи с выполнением рекомендаций Центра, постоянным наблюдением специалистов, мониторингом данных клинико-инструментального исследования резко не выражены. Действительно, НМЗ в настоящее время неизлечимы, но благодаря разработанным схемам медико-социальной реабилитации мы существенно улучшаем качество жизни таких больных. После окончания курса лечения в Центре способствуем их поступлению в вузы, профтехучилища, обучению их различным профессиям.
Таким образом, для достижения наибольшего положительного эффекта в лечении больных с нейромышечной патологией необходима, во-первых, правильная и как можно более ранняя диагностика, а во-вторых — отбор доброкачественных форм, при которых возможно задержать прогрессирование, подобрав адекватную терапию для каждого конкретного случая. При злокачественных формах мы можем только поддержать с помощью лечебных мероприятий функцию сердечной и дыхательной систем и в определенной степени сдерживать прогрессирование болезни. Приводим несколько примеров, подтверждающих вышеприведенный тезис.
Больной П., 16 лет, в 1998 году неврологами Центра был поставлен диагноз: ПМД Дюшенна. Диагноз подтвержден в Донецком медико-генетическом центре. Ребенок ежеквартально проходит курсы реабилитационного лечения в Центре. Амбулаторно получает предписанное медикаментозное лечение. Ребенок сохраняет самообслуживание, ходит самостоятельно, активно занимается программированием, готовится к поступлению в вуз.
Больной С., 18 лет, диагноз: ПМД Эмери — Дрейфуса, в Центре лечится с 2000 года — будущий декоратор.
Больная Е., 17 лет, диагноз: спинальная амиотрофия, 2-й тип, в Центре лечится с 2001 года — пишет прекрасные стихи, прозу.
Больная Г., 19 лет, диагноз: невральная амиотрофия, 1-й тип, в Центре лечится с 1997 года — будущий медицинский психолог.
При достижении 18 лет больные с нейромышечной патологией продолжают поддерживать связь по телефону, Интернету, почте с сотрудниками Центра нейрореабилитации.
Литература
1. Бадалян Л.А. Детская неврология. — М.: Медицина, 1984. — 571 с.
2. Вельтищев Ю.Е., Темин П.А. Наследственные болезни нервной системы. — М.: Медицина, 1998. — 495 с.
3. Горбунова В.Н., Савельева Е.А., Красильников В.В. Молекулярная неврология. — СПб.: Интермедика, 2000. — Ч. 1. — 318 с.
4. Гринио Л.П., Агафонов Б.В. Миопатии. — М.: Медицина, 1997. — 213 с.
5. Евтушенко С.К., Садеков И.А. Наследственные заболевания и пороки развития нервной системы в практике невропатолога, — Донецк: Лебедь, 1994. — 122 с.
6. Иллариошкин С.Н., Иванова-Смоленская И.А. Молекулярные основы прогрессирующих мышечных дистрофий // Журнал неврол. и психиатрии. — 1998. — № 10. — С. 55-62.
7. Казаков В.М. Клинико-молекулярно-генетическая классификация мышечных дистрофий (научный обзор с комментариями) // Неврол. журнал. — 2001 — № 3. — С. 47-52.
8. Крись-Пугач А.П., Гук Ю.М., Зима А.М. Ортопедичні прояви міопатій // Вісник ортопедії, травматології та протезування. — 2004. — № 2. — С. 86-88.
9. Страхова О.С., Белозерова Ю.М., Темин П.А. Кардиомиопатия при прогрессирующей мышечной дистрофии Дюшенна (собственные исследования и обзор литературы) // Интернет.
10. Тетенев Ф.Ф., Бодрова Т.Н., Емельянова Н.В. Биомеханика дыхания у больных с прогрессирующими мышечными дистрофиями // Журн. неврол. и психиатрии. — 2000. — № 8. — С. 38-41.
11. Шишкин С.С., Шаховская Н.И., Крахмалева И.Н. Клинический полиморфизм, генетическая гетерогенность и проблемы патогенеза первичных миопатий // Журн. неврол. и психиатрии. — 2002. — № 2. — С. 54-60.
12. Яхно Н.Н., Штульмен Д.Р., Мельничук П.В. Болезни нервной системы: В 2 т. — М.: Медицина, 2001. — Т. 1. — 743 с.
13. Ahn A.H., Kunkel L.M. The structural and functional diversity of dystrophin // Nature Genet. — 1993. — Vol. 3. — P. 283-291.
14. Burghes A.H., Logan C., Hu X. et al. A cDNA clone from the Duchenne / Becker musculare dystrophy gene // Nature — 1987. — Vol. 328. — P. 434-436.
15. Clemens P.R., Caskey C.T. // Eur. Neurol. — 1994. — Vol. 34. — P. 181-185.
16. Fukuyama Y., Ozawa M. Congenital muscular dystrophy; cliniconosological aspects // Muscular Dystrophy / Ed. S. Ebashi. — Tokyo, 1982. — P. 399-424.
17. Hodgston S.V., Boswinkel E., Walker A. et al. Linkage analysis using nine DNA polymorphisms along the length of the X chromosome locates the gene for Emery-Dreifuss musculare dustrophy to distal Xq (abstract) // J. Med. Genet. — 1986. — Vol. 23. — P. 169-170.
18. Huard J., Bouchard J.P., Roy R. et al. Muscle Nerve. — 1992. — Vol. 15. — P. 550-560.
19. Kazakov V.M., Bogorodinsky D.K., Znoyko Z.V., Skorometz A.A. The facio-scapulo-limb (or the facioscapulohumeral) type of muscular dystrophy. Clinical and genetic study of 200 cases // Eur. Neurol. — 1974. — Vol. 11. — P. 236-260.
20. Kazakov V.M., Rudenko D.I. Clinical variability of facioscapulohumeral muscular dystrophu in Russia // Myscle and Nerve. — 1995. — Suppl. 2. — P. 85-95.
21. Koenig M., Beggs A.H., Moyer M. et al. The molecular basis for Duchenne versus Becker muscular dystrophy: correlation with type of deletion // Am. J. Hum. Genet. — 1989. — Vol. 45. — P. 498-506.
22. Koenig M., Monaco T., Kunkel L.M. The complete sequens of dystrophin predicts a rod-shaped cytoskeletal protein // Cell. — 1988. — Vol. 53. — P. 219-228.
23. Koenig M., Hoffman E.P., Bertolsin C.J. et al. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DND gene in normal and affected individuals // Cell. — 1987. — Vol. 50. — P. 509-517.
24. Mohire M.D., Tandan R., Fries T.J. et al. Neurology. — 1988. — Vol. 38. — P. 573-580.
25. Mueller R., Young I.D. Emery’s elements of medical genetics: 9-th Ed. — Edinburgh, 1995. — P. 72-73.
26. Neuromuscular disorders: gene location // Neuromusc. Disord. — 1999. — Vol. 9, № 5. — P. 1-8.
27. Read L., Galasko C.S. Delay in diagnosing Duchenne muscular dystrophy in orthopaedic clinics // J. Bone Jt. Surg. — 1986. — Vol. 68-B, № 3. — P. 481-482.
28. Taratuto A.L., Lubienski F., Diaz D. et al. Merosin-deficiens congenital muscular dystrophy associated with abnormal cerebral cortical gyration: an autopsy study // Ibid. — 1999. — Vol. 9. — P. 86-94.
29. Tbacher M., Hui J.H.P., Wong H.K. Spinal fusion and instrumentation for paediatric neuromuscular scoliosis: Retrospective revieu // Orthop. Surg. — 2002. — Vol. 10, № 2. — P. 130-137.