Экспериментальный препарат антисмысловой терапии, предназначенный для замедления прогрессирования наследственной формы бокового амиотрофического склероза (БАС, болезнь Лу Герига), доказал свою безопасность в ходе первой фазы клинических испытаний, отчет о которых опубликован в журнале Lancet Neurology.
БАС — медленно прогрессирующее, неизлечимое дегенеративное заболевание центральной нервной системы, при котором происходит поражение как верхних, так и нижних двигательных нейронов, что приводит к параличам и последующей атрофии мышц. Смерть наступает от инфекций дыхательных путей или отказа дыхательной мускулатуры. До сих пор на рынке существует лишь один лекарственный препарат, замедляющий прогрессирование БАС, рилузол, но он имеет весьма ограниченный эффект.
Примерно у 5-10 процентов заболевших — наследственная форма БАС. Около 13 процентов пациентов с генетически обусловленной формой заболевания являются носителями мутаций гена SOD1, кодирующего антиоксидантный фермент супероксиддисмутазу 1.
Специалисты из Washington University School of Medicine (Сент-Луис) и Калифорнийского университета (Сан-Диего) разработали и успешно испытали на животной модели препарат антисмысловой терапии ISIS 333611, который, будучи введенным непосредственно в полость позвоночного канала, снижает концентрацию белка SOD1 в спинномозговой жидкости и тем самым замедляет прогрессирование болезни.
Антисмысловая терапия основана на остановке синтеза ключевого белка, ответственного за развитие заболевания, путем подавления экспрессии соответствующего гена. Это достигается путем введения специально разработанных коротких нуклеотидных последовательностей (антисмысловых РНК, ас-РНК), комплементарных матричной РНК белка-мишени. Управление по продуктам и лекарствам США (FDA) в январе 2013 года одобрило к применению на территории страны препарат антисмысловой терапии Kynamro для лечения очень редкого генетического заболевания, гомозиготной семейной гиперхолестеринемии.
В первой фазе проводившихся на базе Massachusetts General Hospital клинических испытаний антисмыслового олигонуклеотида ISIS 333611, призванной оценить его безопасность и переносимость, приняли участие 32 пациента с БАС, связанным с мутацией SOD1. 24 из них получали ISIS 333611, а остальные — плацебо. Препараты вводились в позвоночник больных постепенно увеличивающимися дозами в течение 11 часов.
В результате специалисты не увидели существенной разницы в выраженности побочных эффектов, в основном связанных со спинальной пункцией, у контрольной группы и группы, получавшей препарат. Анализ образцов спинномозговой жидкости, взятой у пациентов, которым вводили ISIS 333611, сразу после инъекций, показал присутствие в ней препарата.
По результатам первой фазы испытаний ISIS 333611 был признан хорошо переносимым и безопасным. Теперь предстоит следующая фаза клинических испытаний препарата, в ходе которой дозы лекарства будут повышены.
Авторы не исключают, что в будущем ISIS 333611 может быть применен и при ненаследственной форме БАС, если выяснится, что она также связана с мутацией SOD1. Кроме того, полагают авторы, ISIS 333611 может оказаться эффективен и при других генетически обусловленных нейродегенеративных заболеваниях, например, болезнях Альцгеймера, Паркинсона, Хантингтона и других.
Источник: http://medportal.ru
Справка:
Боковой амиотрофический склероз
Боковой амиотрофический склероз (болезнь двигательных нейронов) — хроническое прогрессирующее заболевание нервной системы с избирательным поражением центральных и периферических двигательных нейронов и характеризуется нарастающей слабостью бульбарных мышц, плечевого и тазового пояса, туловища и мышц живота с относительно редким поражением глазодвигательных мышц и сфинктеров тазовых органов.
Эпидемиология. Боковой амиотрофический склероз обычно встречается спорадически, изредка имеются семейные случаи. Частота его от 1,5 до 5 на 100 000 населения, и несколько чаще — у жителей острова Гуам и Марианских островов.
Заболевают в любом возрасте, чаще от 50 лет (семейные случаи) до 65 лет (спорадические случаи). Мужчины болеют несколько чаще (1,4:1).
Этиология и патогенез. Этиология заболевания неизвестна. Предполагается, что оно вызывается вирусом (энтеровирусом, ретровирусом ВИЧ) и протекает по типу медленной инфекции. Об этом свидетельствуют обнаруженные у больных БАС аутоиммунные нарушения, в частности миелинотоксические (антиганглиозидные) антитела в сыворотке крови. Однако существует мнение, что боковой амиотрофический склероз представляет собой гетерогенную группу заболеваний. Семейные случаи (5-10%) аутосомно-доминантным типом наследовапия, нарушается хромосома 21q22.1. Спорадические случаи (90-95%) считаются вирусными.
Патоморфология. Макроскопически головной и спинной мозг выглядят нормальными. Отмечается лишь атрофия прецентральных извилин. Микроскопически в коре мозга определяется уменьшение числа пирамидных клеток, их хроматолиз, шаронидная форма, нейронофагия. В передних рогах спинного мозга обнаруживаются также дегенеративные изменения в нейронах, их гибель, пролиферация астроцитарной глии. Обычно поражаются также двигательные ядра V, VII, X, XI и XII пар черепных нервов в стволе мозга. Параллельно дегенеративным изменениям в телах центральных и периферических мотонейронов отмечается демиелинизация пирамидных систем на всем протяжении (на уровне ствола мозга и боковых канатиков спинного мозга). Патогенез поражения мотонейронов недостаточно выяснен. Можно предположить, что вирус нарушает геном мотонейронов и ускоряются факторы запрограммированной гибели клетки (апоптоза). Наблюдают аутоиммунное воздействие на мотонейроны антител IgG против L-типа кальциевых каналов; избыток свободных радикалов, вызывающих мутацию генов (медь-цинк супероксидазы дисмутазы) с изменением аэробного метаболизма нейронов; повышенную активацию глутаматных рецепторов, что приводит к эксайтотоксичности и избыточному притоку в клетку по натриевым и кальциевым каналам, нарушая активность многих энзимов, вызывая распад белков и липидов с образованием свободных радикалов.
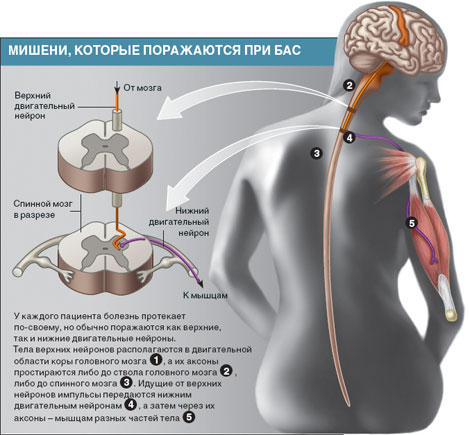
Клиническая картина. Медленное незаметное начало слабости дистальных отделов конечностей или затруднения речи. Сами пациенты или врач обнаруживают при этом атрофии и парезы мелких мышц дистальных сегментов рук и фасцикуляции в этих (и других) мышцах. Фасцикуляции могут быть наиболее ранним симптомом заболевания. Парезы и атрофии постепенно прогрессируют и распространяются на мышцы плечевого пояса, спины, грудной клетки. На первых этапах клинического проявления болезни (у 70%) парезы и атрофии могут быть асимметричными. В последующем наблюдается достаточная симметричность признаков поражения центрального и периферического двигательных нейронов.
Наряду с симптомами периферического пареза выявляются признаки поражения пирамидной системы (высокие глубокие рефлексы на руках и ногах, расширение их рефлексогенных зон, патологические сгибательные кистевые и стопные рефлексы – Россолимо-Вендеровича, Бехтерева, Бабинского). Тонус паретичных мышц может быть повышен, однако при преобладании явлений периферического пареза он бывает низким. В поздней стадии явственно выступают признаки поражения периферического мотонейрона (атрофия мышц, гипо- или арефлексия, фасцикулярные подергивания в них). Боли, парестезии и другие варианты нарушения функции чувствительных нейронов при боковом амиотрофическом склерозе не наблюдаются и возможны только в случаях сочетанной патологии (наиболее часто при сопутствующих спондилогенных нервно-мышечных синдромах).
Для бокового амиотрофического склероза характерно поражение ядер черепных нервов в стволе мозга (IX-XII пар). Появляются и постепенно нарастают расстройства артикуляции, глотания, фонации. Движения языка ограничиваются, определяется его атрофия (уменьшение в объеме, складчатость слизистой, фасцикулярные подергивания в мышцах языка). Мягкое небо свисает, исчезает глоточный рефлекс, больные поперхиваются при приеме жидкой пищи и затрудняются проглатывать плотные фрагменты пищи (хлеб и др.). Выраженное слюнотечение вследствие нарушения автоматического проглатывания слюны, больные постоянно собирают слюну в салфетку, носовой платочек. Из-за слабости мышц шеи голова часто свисает, движения ее ограниченны. Постепенно слабеют жевательные и мимические мышцы вследствие поражения ядер V и VII пар черепных нервов. Лицо становится амимичным, грустным, нижняя челюсть отвисает, жевание затруднено. Двустороннее поражение корково-ядерных трактов приводит к появлению псевдобульбарных симптомов в виде выраженных рефлексов орального автоматизма, непроизвольного плача и смеха.
В цереброспинальной жидкости патологии обычно не выявляется, хотя в четверти случаев умеренно повышено содержание белка. На электромиограмме отмечаются ритмичные потенциалы фибрилляции с амплитудой до 300 мкВ и частотой 5-35 Гц («ритм частокола»). При биопсии мышцы обнаруживают признаки ее денервации.
Течение. В большинстве случаев заболевание начинается в 40-50 лет, однако может начинаться и в более молодом возрасте. В зависимости от преимущественной локализации патологического процесса в начальной стадии выделяют следующие формы бокового амиотрофического склероза: бульварную (у 10%), шейно-грудную, пояснично-крестцовую. Поражение передних рогов и пирамидных систем может быть выражено равномерно. У 10% доминирует поражение периферических мотонейронов. Реже превалируют признаки нарушения функции пирамидной системы при незначительном поражении передних рогов.
Течение заболевания неуклонно прогрессирующее. При наиболее распространенной шейно-грудной форме болезнь принимает восходящий и нисходящий характер. Если заболевание начинается с тазового пояса, то в дальнейшем оно приобретает восходящий тип. При любом варианте начала болезни неизбежно присоединяется бульбарный парез, резко ухудшающий прогноз.
Диагноз и дифференциальный диагноз. Диагноз бокового амиотрофического склероза основывается на сочетании типичных признаков одновременного поражения центрального и периферического мотонейронов в головном и спинном мозге. Характерно отсутствие нарушений чувствительности, координации, функций тазовых органов, патологических изменений в цереброспинальной жидкости, неуклонное прогрессирование заболевания. Важную информацию дает электромиография, которая подтверждает поражение клеток передних рогов спинного мозга.
Дифференцировать боковой амиотрофический склероз следует со спондилогенной шейной миелоишемией, для которой характерны признаки поражения спинномозговых корешков (боль, онемение, трофические расстройства) и рентгенологические находки выраженного остеохондроза, спондилоартроза, грыжи дисков, сужение позвоночного канала. В клинике спондилогенной шейной миелоишемии доминируют симптомы поражения двигательных структур исключительно шейного утолщения и отсутствуют бульбарные расстройства. Окончательный диагноз в сомнительных случаях устанавливается после миелографии и МРТ шейного отдела позвоночника. При прогредиентных формах клещевого энцефалита следует учитывать анамнез, эндемичность заболевания, более длительное доброкачественное течение, высокий титр специфических антител в крови.
Для сирингомиелии, проявляющейся также дистальными атрофиями, типичны длительное течение, болевой синдром, расстройства чувствительности диссоциированного типа, вазомоторные и трофические нарушения, признаки дизрафичного статуса.
В некоторых случаях боковой амиотрофический склероз необходимо дифференцировать со спинальным амиотрофическим процессом сифилитической этиологии, для которого характерны медленное течение, корешковые боли, практически отсутствие бульбарных симптомов, сочетание со зрачковыми феноменами (синдром Аргайла Робертсона), положительная реакция Вассермана в крови и цероспинальной жидкости, наличие в ней плеоцитоза.
Клиническая картина, сходная с боковым амиотрофическим склерозом, может быть обусловлена рецессивной бульбарной атрофией Кеннеди, спинальной мышечной атрофией, энцефаломиелопатией при висцеральных карциноматозных процессах, рассеянном склерозе, болезни Пика и др.
Лечение. Требуется мультидисциплинарное ведение больных (невролог, семейный врач, физиотерапевт, методист по лечебной физкультуре, логопед, медицинская сестра, социальный работник). Эффективной терапии пока нет. Отсрочить летальный исход на несколько месяцев способен рилузон. Его механизм действия связан с торможением высвобождения из нейронов возбуждающей аминокислоты глутамата, которая запускает процесс дегенерации нейронов. Показаны метаболические препараты: витамин Е, витамины группы В, АТФ, ноотропы, кортексин, анаболические гормоны (ретаболил 1 мл в/м, 1 раз в неделю), L-карнитин, глицин, факторы роста нервов. Рекомендуют введение трипептида тиреотропин-рилизинг-гормона в больших дозах внутривенно и в малых дозах длительно подкожно или внутримышечно. Возможно применение также дериватов данного гормона. Эти препараты являются нейромодуляторами произвольной двигательной системы, поэтому целесообразно широкое их применение при болезнях мотонейрона. Для улучшения нервно-мышечной проводимости назначают дибазол, оксазил, прозерин, для уменьшения спастичности, особенно в нижних конечностях, — мидокалм, сирдалуд. При крампи назначают фенитоин. В начальный период заболевания до развития выраженных атрофии показан легкий массаж конечностей. При слюнотечении применяют атропин, гиосцин и т.п.
Лечение проводят курсами по нескольку раз в год. При развитии тяжелых бульбарных нарушений (невозможность глотания) необходимо кормление через зонд, парентеральное введение жидкостей.
Прогноз. Тяжесть состояния больных определяется бульбарными и дыхательными расстройствами. В отношении жизни прогноз неблагоприятен. Длительность заболевания от 2 до 10 лет и зависит от варианта развития болезни. Наиболее неблагоприятная форма — бульбарная, приводящая к летальному исходу через 1,5-2 года. Смерть наступает вследствие паралича дыхательного центра, интеркуррентных инфекций, истощения. Шейно-грудная форма протекает от 4 до 8 лет, пояснично-крестцовая — 8-10 лет. При присоединении бульбарных нарушений пациенты редко живут более 2 лет.
Источник: http://all-about-genetics.ru