


Миодистрофия Дюшенна — тяжелое наследственное заболевание, которым болеют только мальчики. Первые проявления болезни видны уже в первый год…
Продолжить чтениеМетка: пропуск экзона
Новое лечение может улучшить состояние 45 % пациентов с мышечной дистрофией Дюшенна
Новый препарат, разработанный под руководством профессора медицинской генетики Тосифуми Йокоты из Университета Альберты, может быть полезен…
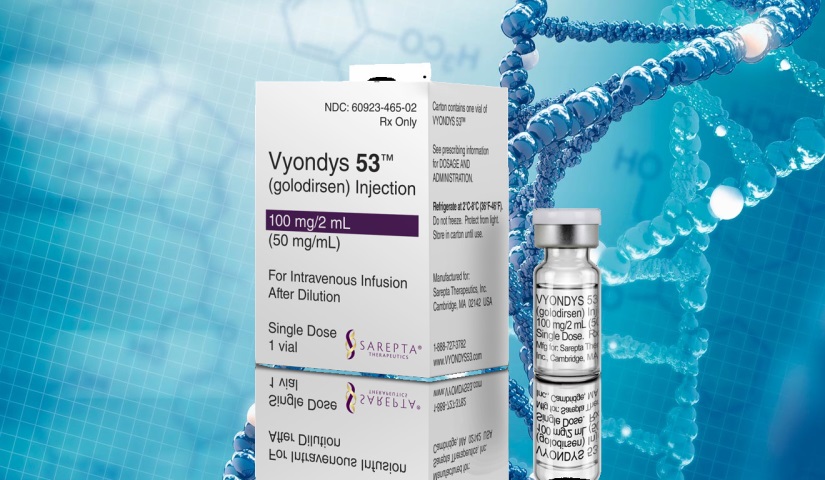
FDA предварительно одобрило таргетный препарат компании Sarepta для лечения МДД
FDA одобрило по ускоренной процедуре Vyondys 53 – второй препарат для лечения мышечной дистрофии Дюшенна (МДД)…
Продолжить чтение
«Миоредактирование» — новый подход к применению CRISPR/Cas9 в терапии миодистрофии Дюшенна.
Миодистрофия Дюшенна (МДД) – прогрессирующее мышечное дегенеративное заболевание, одно из самых распространенных смертельных генетических заболеваний (распространенность…
Продолжить чтение
FDA одобрило первый препарат для лечения мышечной дистрофии Дюшенна
В США одобрено первое лекарство для лечения миодистрофии Дюшенна. Это заболевание поражает мальчиков. Первые симптомы появляются…
Продолжить чтение
В США одобрен первый препарат для лечения мышечной дистрофии Дюшенна
Управление по контролю за пищевыми продуктами и лекарственными средствами США (Food and Drug Administration — FDA) одобрило первый инъекционный препарат…

Метод редактирования РНК эффективен при лечении тяжелой формы мышечной дистрофии
Предварительные результаты использования методики редактирования РНК, называемой «пропуском экзона», полученные учеными из Northwestern Medicine (США) и Университета…
Продолжить чтение
Первое изучене NS-065 / NCNP-01, антисмысловых морфолино олигонуклеатидов для пропуска экзона 53 при мышечной дистрофии Дюшенна
В настоящее время проходит фаза 2/3 клинического испытания пропуска экзона 51 при мышечной дистрофии Дюшенна (МДД),…
Продолжить чтение
BioMarin завершает представление информации для ускоренного одобрения дрисаперсена для лечения мышечной дистрофии Дюшенна.
Заявление сотрудника научной программы АМД Лауры Хагерти: «Это очень обнадеживающие новости для сообщества МДД, и мы…

Системное введение PRO051 при мышечной дистрофии Дюшенна.
Местное внутримышечное введение антисмыслового олигонуклеотида PRO051 у пациентов с мышечной дистрофией Дюшенна с соответствующими мутациями,как сообщалось…
Продолжить чтение
Сарепта подтверждает приверженость развитию етеплирсена и другим препаратам для пропуска экзона
Камбридж, штат Массачусетс. Сарепта, разработчик етеплирсена и других препаратов для пропуска экзона, предназначенных для лечения мышечной…
Продолжить чтение
Дрисаперен для лечения мышечной дистрофии Дюшенна
Дрисаперен представляет собой 2′-O-метил-фосфоротиоат антисмысловой олигонуклеотид, который вызывает пропуск экзона 51, чтобы исправить мутацию мышечной дистрофии…
Продолжить чтение
Разработка лекарственных средств для МДД 2014
Разработка лекарственных средств для мышечной дистрофии Дюшенна (МДД) идет по нескольким направлениям. Вот некоторые обновления на…
Продолжить чтение
Фаза 3 исследования етеплирсена
Мальчики с мышечной дистрофией Дюшенна в возрасте от 7 до 16 лет в настоящее время привлекается…
Продолжить чтение
Sarepta Therapeutics опубликовала новые данные по своим исследованиям.
Sarepta Therapeutics является разработчиком етеплирсена и других соединений, которые нацелены на конкретные разделы («экзоны») в гене…
Продолжить чтение
Sarepta будет стремиться к ускоренному утверждению Этеплирсена (Eteplirsen) и к проведению дополнительных исследований
Sarepta Therapeutics, разработчик экспериментального лекарственного препарата Этеплирсена, предназначенного для мышечной дистрофии Дюшена (МДД), сообщает, что они…
Продолжить чтение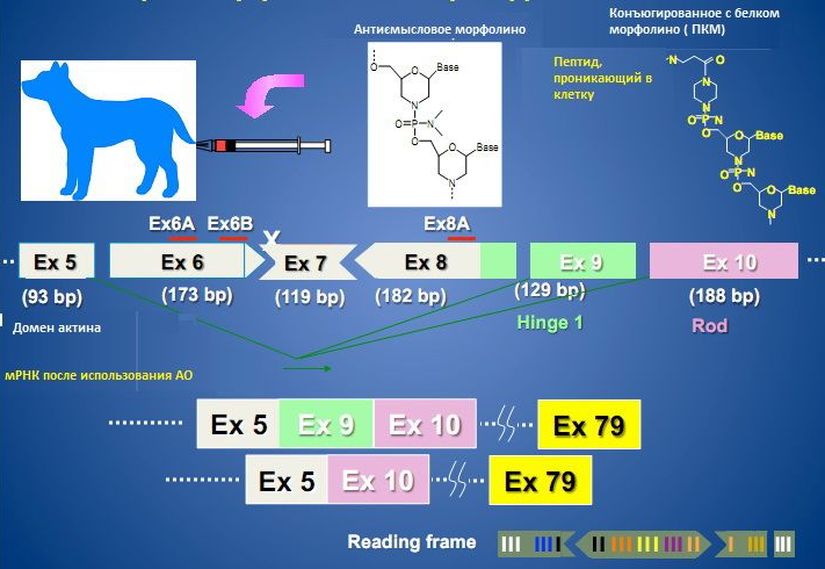
Пропуск экзона
Перевод презентации одного из докладов, представленных на Конференции по формированию связей. Доклад посвящен развитию метода пропуска…
Компания Prosensa для МДД сообщества организует корректировку программы по пропуску экзонов
18 ноября 2013 года в ходе проведения пресс-релиза и конференц-звонка, содержание которого уже отправлено в архив,…
Продолжить чтение