


Написать эту статью меня подтолкнул шквал публикаций о поиске 2 миллионов долларов на лечение обреченных детей со спинальной мышечной атрофией.

Почему сейчас это редкое заболевание – спинальная мышечная атрофия- стало так часто упоминаться в СМИ? Что, раньше им не болели? Болели и известно оно с конца 19 века, когда немецкий врач Гидо Вердниг и австрийский Иоганн Хоффман описали это тяжелое заболевание. Самое интересное, что уже первые случаи заболевания были описаны в одной семье, и это несомненно наталкивало врачей на мысль о генетических причинах заболевания. Но развитие генетики в конце 19 века явно не позволяло делать какие-то выводы.
Что это за болезнь – спинальная мышечная атрофия?
Я не собираюсь детально описывать это тяжелое заболевание, его детальное описание есть в Интернете.
Если говорить коротко, за счет мутации в определенном гене ( если точно – SMN1) отмечается нарушение мотонейронов спинного мозга и как следствие атрофия мышц. Есть несколько форм этого тяжелого заболевания. При самых тяжелых формах ребенок погибает до 2-х лет, при менее тяжелых – выживает, но обречен быть инвалидом-«колясочником», зачастую требующим дыхательной поддержки с помощью аппарата ИВЛ. Самое трагичное, что у выживших пациентов не страдает интеллект и список известных людей со спинальной мышечной атрофией достаточно представительный. А доживший до подросткового возраста ребенок выглядит вот так
А при чем здесь генетика?
Это заболевание относится к громадной группе заболеваний с аутосомно-рецессивным наследованием. Есть единый каталог всех описанных врачами всего мира наследственных заболеваний OMIM (расшифровывается Online Mendelian Inheritance in Man, omim.org). Там описано невероятное количество болезней, и подавляющее их большинство именно аутосомно-рецесивного типа наследования. Давайте посмотрим на схеме этот звучащий таинственно и интригующе тип наследования.
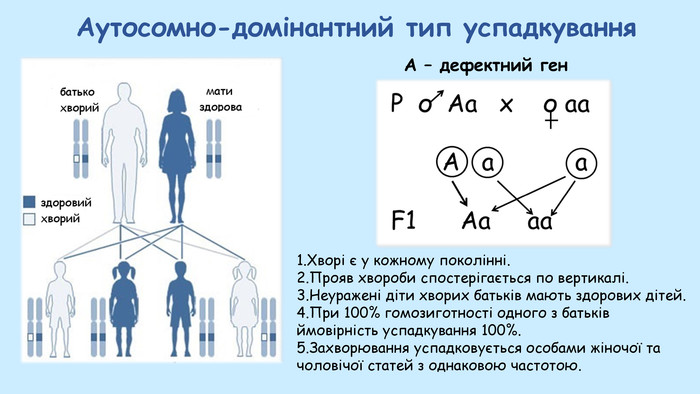
Кстати, взята эта схема из школьного онлайн курса по биологии для 10-го класса. Да-да, все , кто по крайней мере имеет среднее образование, это учил в школе. Если не ошибаюсь, эту тему проходят весной. Весна, 10 класс, 16 лет – ну разве кто-то это запомнит? Но факт остается фактом – это учат в школе.
Какие самые частые заболевания наследуются по этому типу? Это уже упомянутая спинальная мышечная атрофия, частота примерно 1на 6000 новорожденных. Но еще более частое заболевание – муковисцидоз, частота 1 случай на 2500 новорожденных. Кстати, муковисцидоз – крайне тяжелое и неизлечимое заболевание, 90% пациентов не доживают до 2-х лет.
Справедливости ради, там же , в школьном курсе биологии учат и Х-сцепленные заболевания, которыми болеют только мужчины, а носителями мутантного гена являются женщины.

По этому типу наследуется всем известная гемофилия, склонность в «несвертываемости» крови, всем хорошо известная по истории царской семьи Романовых.
Но давайте вернемся к спинальной мышечной атрофии, которая у всех сейчас на устах. Почему?
Лечение спинальной мышечной атрофии.
Именно это заболевание явилось одним из первых удачных примеров удачного излечения. Сначала в декабре 2016 года был и разрешен к применению в США препарат Спинраза. После долгих дискуссий, он был разрешен к применению в около 40 странах мира. Вводится этот препарат в спинно-мозговой канал ( люмбальная пункция) и лечение этим препаратом обходится 750.000 долларов в первый год лечения и потом 375.000 долларов ежегодно пожизненно . Несмотря на это, лечение этим препаратом покрывается в 40 странах мира. Этот препарат относится к группе одних из самых дорогих в мире. Поэтому его введение в программы покрытия государством сопровождались во многих странах бурными дискуссиями. Так, норвежский парламент назвал этот препарат «неэтично дорогим» и сначала отказался покрывать лечение больных СМА этим препаратом.
В мае 2019 года появился новый препарат для лечения СМА – Золгенсма. Не останавливаясь подробно на сложных механизмах его действия, упомянем лишь его главное лечение от своего предшественника – Спинразы. Золгенсма вводится однократно и внутривенно. Всего одна инъекция, но она стоит ( внимание, сядьте!) 2,1 миллиона долларов. Да-да, это не ошибка и не опечатка. Именно такая умопомрачительная цена позволила этому препарату занять место в Книге рекордов Гинесса как самому дорогому лекарственному средству.
Вопрос о победе над СМА — это не только деньги. Какие проблемы еще стоят перед пациентами , их родителями и врачами?
- Заболевание надо выявить как можно раньше. СМА может проявится с рождения, но есть варианты и более позднего проявления этого коварного заболевания, в 6-12 месяцев и позже. Поэтому сейчас стоит вопрос о включении в скрининг ( тотального обследования) новорожденных и этого заболевания. Просто хочу напомнить, что в Украине всех новорожденных обследуют ( или должны обследовать) на 4 заболевания. В мире уже этот список расширен до 40 заболеваний. Включение СМА в этот список еще больше удорожает скрининг новорожденных. Для ориентира — Для Украины на ближайшие десятилетия скрининг новорожденных на СМА и последующее лечение малореальны.
- Наилучший эффект лечения по имеющимся данным достигается при начале лечения до 2-х месяцев. Для Золгенсмы этот срок, начала лечения, 7 месяцев. Если обобщить имеющиеся данные, то начинать лечение можно и до 2-х лет, но эффект будет скромнее. Ребенок может начать сидеть, в редких случаях стоять, иногда – ходить с поддержкой. Для измученных родителей и это победа, но абсолютно здоровым ребенок так и не станет.
- Лечение этими препаратами не лишено определенных осложнений, иногда достаточно тяжелых.
- Ну и опять же деньги, пресловутые деньги. Где их взять, если государство не может помочь?
Что делать?
Единственный эффективный механизм избежать рождения ребенка больного Спинальной Мышечной Атрофией – это обследование всех людей, собирающихся обзавестись потомством, на носительство гена SMN1. Для того чтобы посчитать эффективность этого подхода, необходимо иметь знания в математике и биологии на уровне средней школы.
Итак, носителем этого гена является 1 из 40 граждан Украины, независимо от пола. Вероятность того, что встретятся 2 носителя гена ( хотел написать образ из песни «два одиночества») — 1/40*1/40=1/1600. И при этой «встрече» вероятность рождения больного ребенка = 1/1600*1/4= 1/6400. Почему ¼ ? А вспомните уже упомянутую программу по общей биологии 10-го класса средней школы. И сразу все станет понятно.
Хочется разбавить сухие цифры литературной классикой. «Мы все учились понемногу чему-нибудь и как-нибудь» — это ироничный Пушкин А.С. А может из советской пропаганды – «Знание-сила»? Вооруженные силой знаний мы легко посчитаем, что в Украине за 2020 год родилось 293.000 детей. При частоте СМА 1 случай на 6400 родов, должно было родиться 293.000/6400= 46 детей. Сколько их родилось на самом деле , мне кажется, не знает никто. Да и многим маленьким пациентам диагноз еще не поставлен.
Давайте пофантазируем. Если обследовать всех 293.000 женщин, которые родили в 2020 году перед наступлением беременности или хотя бы в ранние сроки ( до 10-12 недель), то будет выявлено 293.000/40 ( частота носительства гена в нашей популяции), то будет выявлено 7325 носителей гена. Следующим этапом необходимо обследовать их мужей. Должно быть выявлено 7325/40=183 супружеские пары с высоким риском рождения ребенка со СМА. Что им делать?
У этих пар есть следующий выбор:
А) провести предимплантационную генетическую диагностику, а по-простому – ЭКО с генетическим исследованием эмбриона;
Б) провести пренатальную диагностику, т.е. исследование плода во время беременности и не допустить рождения больного ребенка.
Как показывает уже накопившийся зарубежный опыт, 70-80% супружеских пар с высоким риском, выбирает предимплантационную генетическую диагностику. Остальные – пренатальную диагностику или реже другие способы уберечься от рождения больного ребенка ( вплоть до расторжения брака или отказ от деторождения).
Кстати, если нашу «виртуальную» программу профилактики СМА выразить в финансовом эквиваленте, то в масштабах Украины она бы стоила бы где-то 300 млн. гривен. Соответствующее лечение неминуемо рожающихся детей – примерно 3 млрд. гривен. Учитывая, что у нас не ни профилактики, ни государство пока не покрывает лечение этих детей эти цифры существуют пока лишь в виртуальной реальности.
Что дальше?
По большому счету, то, что я рассказал, известно достаточно давно. Возможность выявлять мутацию в гене СМА, а именно SMN1, появилась в 90-е годы. А в 2008 году обследование на носительство этого гена было рекомендовано Американским Колледжем Акушеров и Гинекологов (The American College of Obstetricians and Gynecologists). Тогда рекомендовали обследование на носительство гена СМА вместе с обследованием на носительство гена муковисцидоза. Эти два заболевания являются наиболее частой причиной детской смертности от наследственных заболеваний. Давайте посмотрим эволюцию скрининга на носительство генетических заболеваний на следующей схеме.
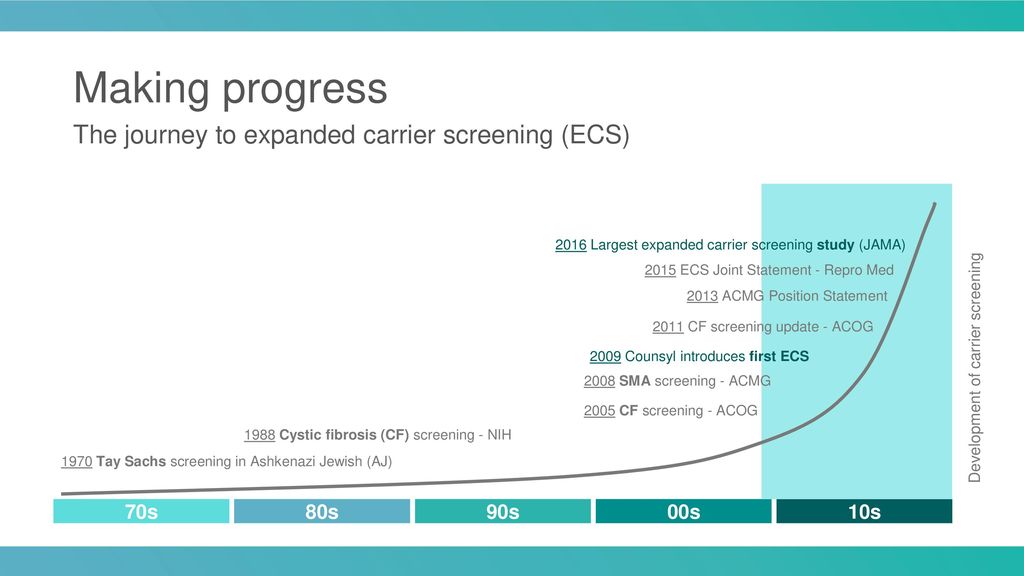
Скрининг на СМА и муковисцидоз появился в мире в середине 2000-х годов, а с 2010 года, с появлением новых технологий, стало возможным одновременное исследование на носительство от 100 до 400 генов . Цель благая – не допустить рождение больного ребенка! В США востребованность скрининговых программ на носительство генов «оживила» политика страховых компаний. Логика их активности проста – оказалось, что гораздо дешевле предусмотреть рождение больного ребенка, чем потом тратить огромные деньги на его лечение.
А что у нас?
А ничего. За генетическими исследованиями обращаются в основном пациенты, у которых уже «что-то случилось». Проспективных, т.е. «профилактических» исследований крайне мало. Почему? Во-первых, не очень наши будущие родители об этом знают и думают. Во-вторых, не развит у нас «страховой менталитет». В-третьих, если в родословной все нормально, то слабо верится попадание в пресловутый «процент риска». Государственных программ по профилактике наследственных заболеваний у нас нет и в ближайшем будущем не предвидится. Надо быть очень большим оптимистом, чтобы представить, что скудно финансируемое украинская медицина сможет обеспечить обследование ВСЕХ БУДУЩИХ РОДИТЕЛЕЙ ИЛИ БЕРЕМЕННЫХ на носительство генов. Поэтому спасение утопающих – дело рук самих утопающих. Можно ли в Украине пройти эти исследования? Можно. Однако затраты на маркетинг во всеукраинском масштабе явно не по плечу ни одной частной клинике или лаборатории. А государственной образовательной программы просто нет. Кстати, опыт многих стран демонстрирует, что проблема не только в финансовом обеспечении программ, а и в информированности населения в жизненной необходимости подобных программ. Да и знания врачей в области клинической генетики в Украине явно далеки от идеала. Наше медицинское образование вопросы медицинской генетики явно проигнорировало.
PS. И все-таки я остаюсь оптимистом. Куча публикаций на сбор средств для лечения детей, родившихся со СМА и другими наследственными заболеваниями так или иначе порождает в обществе желание не попасть в «печальную статистику». И каждая семья, избавленная от трагедии рождения больного ребенка, и каждый нерожденный больной ребенок, будет большой победой разума человека над природой. Ну а кроме этого, это обследование будет явно несоизмеримо дешевле 2млн. долларов на лечение больного ребенка…
Похожие статьи:

Спасительные заблуждения. Мифы, во власти которых часто находятся родители тяжелобольных детей

Спинальная мышечная атрофия (СМА): ответы на самые часто задаваемые вопросы
«Есть лекарство, болезнь и ребенок. Всё!». Врач Василий Штабницкий — о том, почему государство даже не знает, сколько в стране больных СМА